Emergence and spread of SARS-CoV-2 P.1 (Gamma) lineage variants carrying Spike mutations 𝚫141-144, N679K or P681H during persistent viral circulation in Amazonas, Brazil
Felipe Naveca 1, Valdinete Nascimento 1, Victor Souza 1, André Corado 1, Fernanda Nascimento 1, George Silva 1, Matilde Mejía 1, Ágatha Costa 1, Débora Duarte 1, Karina Pessoa 1, Maria Júlia Brandão 1, Michele Jesus 2, Luciana Gonçalves 3, Cristiano Fernandes 3, Tirza Mattos 4, Roberto Lins 5, Danilo Coêlho 5, Gabriel Luz Wallau 5, Edson Delatorre 6, Tiago Gräf 7, Marilda Mendonça Siqueira 8, Paola Cristina Resende 8, Gonzalo Bello 9
Affiliations:
1 Laboratório de Ecologia de Doenças Transmissíveis na Amazônia, Instituto Leônidas e Maria Deane, Manaus, Amazonas, Brazil.
2 Laboratório de Diversidade Microbiana da Amazônia com Importância para a Saúde, Instituto Leônidas e Maria Deane, Manaus, Amazonas, Brazil.
3 Fundação de Vigilância em Saúde do Amazonas - Dra. Rosemary Costa Pinto, Manaus, Amazonas, Brazil.
4 Laboratório Central de Saúde Pública do Amazonas, Manaus, Amazonas, Brazil.
5 Instituto Aggeu Magalhães, Fundação Oswaldo Cruz, Recife, Pernambuco, Brazil.
6 Departamento de Biologia. Centro de Ciências Exatas, Naturais e da Saúde, Universidade Federal do Espírito Santo, Alegre, Brazil.
7 Instituto Gonçalo Moniz, Fundação Oswaldo Cruz, Salvador, Brazil.
8 Laboratório de Vírus Respiratórios e do Sarampo (LVRS), Instituto Oswaldo Cruz (IOC), Rio de Janeiro, Brazil
9 Laboratório de AIDS e Imunologia Molecular, Instituto Oswaldo Cruz, FIOCRUZ, Rio de Janeiro, Brazil.
Corresponding authors:
Felipe Gomes Naveca
[email protected] ORCID: 0000-0002-2888-1060
Gonzalo Bello
[email protected] ORCID: 0000-0002-2724-2793
Summary
The Amazonas was one of the most heavily affected Brazilian states by the COVID-19 epidemic that resulted in the infection of a large proportion of individuals from that state, particularly during the second wave associated with the rapid spread of the SARS-CoV-2 Variant of Concern (VOC) Gamma (lineage P.1). Despite the large number of infected people, SARS-CoV-2 continues to circulate in the Amazonas state, and an average of 518 new cases per day was diagnosed through 1st May and 15th June 2021. Understanding how SARS-CoV-2 may persist in a human population with high levels of immunity is of paramount importance. To this end, we conducted a genomic survey that analyzed 744 SARS-CoV-2 whole-genome sequences from individuals living in the Amazonas state from 01st January to 31st May 2021. Our study reveals a sharp increase in the prevalence of P.1 variants harboring two types of additional changes in the Spike protein: deletions in the N-terminal (NTD) domain (particularly 𝚫141-144) or mutations at the S1/S2 junction (N679K or P681H). Together, these emergent P.1 plus variants resemble 78% of all SARS-CoV-2 positive cases genotyped in the Amazonas state in the second half of May 2021. These findings reveal that persistent transmission of SARS-CoV-2 VOC Gamma in the Amazonian population has been associated with continuous viral evolution and community spread of a second-generation VOC P.1 that could be more transmissible or resistant to neutralization than the parental one.
Background
The Amazonas was one of the most heavily affected Brazilian states by the COVID-19 epidemic, and by 15th June 2021, 13,162 deaths had been reported [1]. The COVID-19 epidemic in Amazonas was characterized by two waves of exponential growth (Fig. 1a). The first one started in March 2020 and peaked around early May 2020, and was primarily associated with the introduction and dissemination of lineages B.1.195 and B.1.1.28 [2]. The second one started in December 2020 and peaked around early February 2021 and was associated with the local emergence and rapid spread of a new SARS-CoV-2 Variant of Concern (VOC), firstly detected in Japanese travelers returning from the Amazonas State, Brazil [3], designated as lineage P.1 or Gamma [2, 4, 5]. Since mid-February 2021, the number of SARS-CoV-2 deaths dropped and remained roughly stable (7-day average <20) from May to June 2021.
Despite both the number of infected people in the two waves of the COVID-19 pandemic and the percentage (37.1%) of fully vaccinated individuals [6] supports a high prevalence of individuals with anti-SARS-CoV-2 antibodies in the Amazonas state, the virus continues to circulate and an average of 518 new SARS-CoV-2 positive cases per day were diagnosed during 1st May and 15th June 2021 [7]. We hypothesize that continuous transmission of the VOC Gamma in the Amazonian population with high levels of immunity acquired from both natural SARS-CoV-2 infection or vaccination might select for second-generation of P.1 variants that are more resistant to neutralization than the parental virus. To test this hypothesis, we generated 744 SARS-CoV-2 high-quality, whole-genome sequences from individuals living in the Amazonas state from 01st January to 31st May 2021 (Fig. 1a). Viral sequences were generated at FIOCRUZ Amazônia, which is part of both the Amazonas state health genomics network (REGESAM) and the consortium FIOCRUZ COVID-19 Genomics Surveillance Network of the Brazilian Ministry of Health (http://www.genomahcov.fiocruz.br/).
Results and Discussion
Our genomic survey confirms that P.1 was the most prevalent lineage representing 99% (744 of 752 genomes) of all samples analyzed in the Amazonas state across the study period. Furthermore, our results reveal a sharp increase in the prevalence of P.1 variants harboring two types of additional changes in the Spike (S) protein: deletions in the N-terminal (NTD) domain (NTDdel) or mutations at the S1/S2 junction (N679K or P681H) (Fig. 1b). The P.1+NTDdel variants increased from <10% in January-April to 16% in May 2021. NTDdel at the N3 loop (𝚫144, 𝚫141-144, and 𝚫138-143) were much more frequent than that at N5 loop (𝚫241-243 deletion), and the P.1+𝚫141-144 variant displayed the most significant increase in prevalence during May 2021 (Fig. 1c). The P.1+P681H and P.1+N679K variants increased from 0% in the first half of March to 29% and 32% in the second half of May 2021, respectively. Together, the P.1+NTDdel, P.1+P681H, and P.1+N679K variants resemble 78% of all SARS-CoV-2 positive cases that underwent genomic sequencing in Amazonas in the second half of May 2021.
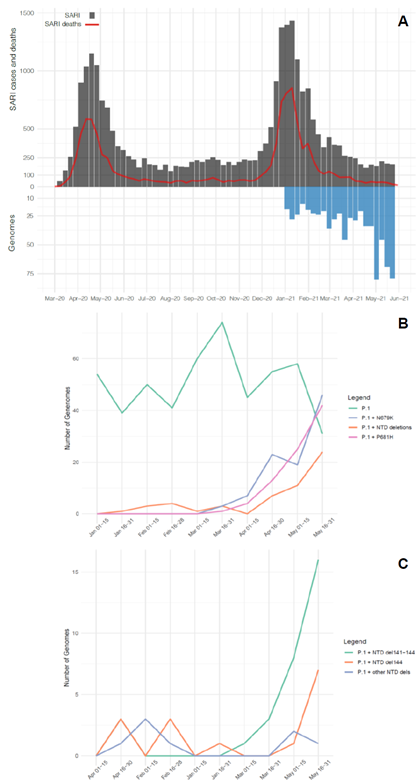
Figure 1. Temporal distribution and genetic diversity of SARS-CoV-2-positive samples from the Amazonas state during 2021. A) Graph depicting the temporal evolution of SARI cases and SARI deaths based on the date of symptom onset (source, http://info.gripe.fiocruz.br) as a proxy for the COVID-19 epidemic curve in Amazonas state, along with the number of SARS-CoV-2 whole-genome sequences generated between January and May 2021. B) Number of genomes corresponding to the different P.1 lineage variants detected. C) Number of genomes corresponding to the different P.1+NTDdel lineages detected.
We combined high-quality Amazonian P.1 sequences generated in this and previous studies with P.1+NTDdel (𝚫144, 𝚫143-144, and 𝚫141-144), P.1+P681H and P.1+N679K sequences detected in other Brazilian states that were available at the EpiCoV database in GISAID (https://www.gisaid.org/) on 25th June 2021. The Maximum Likelihood (ML) phylogenetic analysis supports that the NTDdel at N3 loop arose multiple times during the evolution of lineage P.1 in Brazil, in agreement with previous observations [8]. Moreover, this analysis revealed a highly supported (aLRT = 86%) monophyletic clade designated as P.1+𝚫141-144AM that comprises most (n = 27/31, 88%) Amazonian sequences with that NTD deletion (Fig. 2). Mutation S:N679K arose two times giving rise to a major clade P.1+N679KAM-I (aLRT = 79%) that comprises most sequences (n = 90/98, 92%) from the Amazonas state as well as one sequence detected in Rio de Janeiro state, and a minor clade P.1+N679KAM-II (aLRT = 78%) comprising eight sequences from Amazonas (Fig. 2). Mutation S:P681H also arose at least two times in Brazil, giving rise to lineages P.1+P681HAM (aLRT = 79%) and P.1+P681HBR (aLRT = 74%) that comprise all sequences sampled in Amazonas (n = 85) and other Brazilian states (n = 122), respectively (Fig. 2).
Most sequences belonging to clades P.1+𝚫141-144AM (96%), P.1+P681HAM (78%), P.1+N679KAM-I (79%), and P.1+N679KAM-II (75%) were sampled in the capital city of Manaus between 12th March and 31st May 2021. The P.1+P681HBR sequences were sampled in states from Southern (Paraná and Santa Catarina), Southeastern (Rio de Janeiro and São Paulo), Central-Western (Goiás), Northeastern (Alagoas, Maranhão, and Paraíba), and Northern (Pará and Tocantins) regions between 12th March and 06th June 2021 and its frequency among P.1 infections from outside the Amazonas state increase from 0% in February to 21% in June 2021. The mutational profile was investigated using the Nextclade tool (https://clades.nextstrain.org) revealing a variable total number of lineage-defining mutations in clades P.1+𝚫141-144AM (n = 6), P.1+P681HAM (n = 3), P.1+P681HBR (n = 3), P.1+N679KAM-I (n = 1) and P.1+N679KAM-II (n = 1) (Table 1). It is interesting to note that clades P.1+N679KAM-I and P.1+N679KAM-II displayed the same lineage-defining amino acid substitution, but different lineage-defining nucleotide mutations.
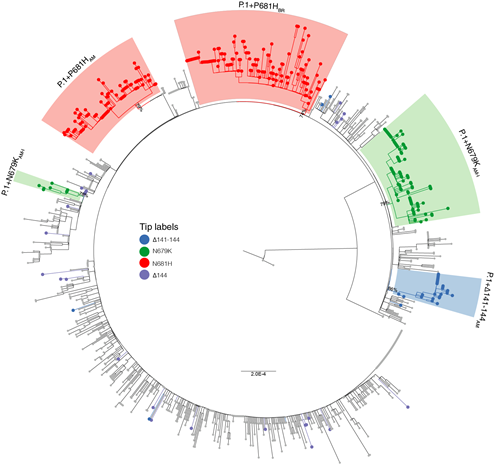
Figure 2. Maximum likelihood (ML) phylogenetic tree of P.1 Amazonian sequences and P.1+NTDdel, P.1+N679K, and P.1+P681H sequences detected outside Amazonas. Major Tips were colored according to the S mutations as indicated in the legend. Major P.1 sub-lineages carrying additional mutations/deletions in the S protein were highlighted with colored boxes. The aLRT support values are indicated in key branches, and branch lengths are drawn to scale, with the bar indicating nucleotide substitutions per site.
Table 1. Lineage-defining mutations present in >95% of P.1 sub-lineage sequences.
| P.1 sub-lineage | Nucleotide | Amino acid |
|---|---|---|
| P.1+𝚫141-144AM | C5526T | ORF1a:T1754I |
| C16193T | ORF1b:P909L | |
| A21979T | - | |
| 𝚫21983-21994 | S:𝚫141-144 | |
| T27826C | ORF7b:M24T | |
| C27942T | ORF8:H17Y | |
| P.1+P681HAM | C10615T | - |
| C15714T | - | |
| C23604A | S:P681H | |
| P.1+P681HBR | C1912T | - |
| C16293T | - | |
| C23604A | S:P681H | |
| P.1+N679KAM-I | T23599G | S:N679K |
| P.1+N679KAM-II | C3117T | ORF1a:T951I |
| A18945G | - | |
| T23599G | S:N679K |
The temporal structure analysis using TempEst [9] revealed that the overall divergence of the new sub-lineages P.1+𝚫141-144AM, P.1+N679KAM, P.1+P681HAM, and P.1+P681HBR is consistent with the overall substitution pattern of other P.1 sequences (Fig. 3a). Bayesian reconstruction using a strict molecular clock model with a uniform substitution rate prior (8 - 10 × 10-4 substitutions/site/year) as implemented in BEAST 1.10.4 [10] estimated the emergence of the sub-lineage P.1+N679KAM at 16th February, 2021 (95% High Posterior Density [HPD]: 16th January - 13th March 2021), sub-lineage P.1+P681HBR on 27th February, 2021 (95% HPD: 02th February - 10th March 2021), sub-lineage P.1+P681HAM at 06th March, 2021 (95% HPD: 09th February - 26th March 2021), and sub-lineage P.1+𝚫141-144AM at 06th March, 2021 (95% HPD: 13th February - 12th March 2021) (Figs. 3b-3e). The city of Manaus was traced as the most probable source location (PSP ⩾ 0.74) of clades P.1+𝚫141-144AM, P.1+N679KAM-I and P.1+P681HAM, while the state of São Paulo (PSP = 0.72) was the most probably epicenter of clade P.1+P681HBR.
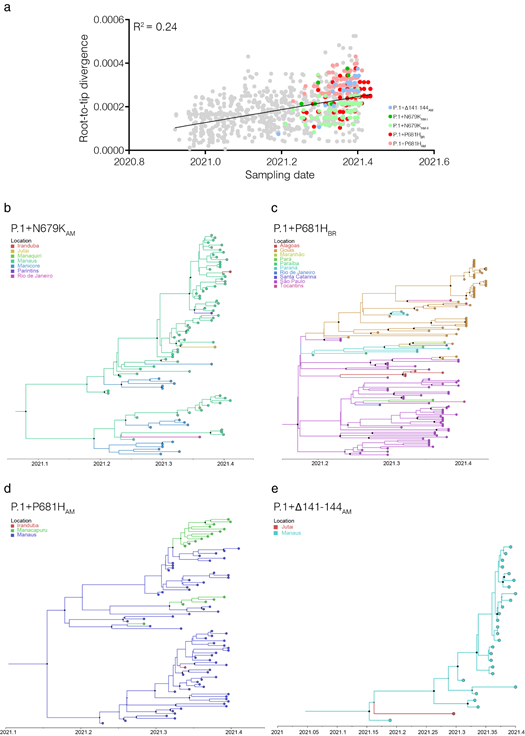
Figure 3. Temporal structure and phylogeographic reconstruction of the P.1+NTDdel, P.1+N679K, and P.1+P681H clades. a) root-to-tip regression of genetic divergence against dates of sample collection. P.1 sequences were colored gray, while each P.1 subclade carrying deletions or additional mutations in S was colored following the legend. Time-resolved maximum clade credibility phylogenies of each P.1 subclade defined in the ML analysis: b) P.1+N679KAM; c) P.1+P681HBR; d) P.1+P681HAM; e) P.1+𝚫141-144AM. Tips and branches colors indicate the sampling location and the most probable inferred location of the nodes, respectively, as indicated in the legend for each tree. Circles at nodes are scaled to the posterior probability. All horizontal branch lengths are time-scaled, and the tree was automatically rooted under the assumption of the strict molecular clock model.
The VOC Gamma is sensitive to neutralizing antibodies (NAb) directed against the NTD antigenic supersite and NTD deletions mapping in the recurrent deletion region-2 (RDR-2), such as the 𝚫141-144, has been shown to confer resistance to those antibodies [11, 12]. Studies of intra-host SARS-CoV-2 evolution revealed that NTD deletion 𝚫141-144 emerged following therapy with convalescent plasma in immuno-compromised hosts [13, 14], during persistent SARS-CoV-2 infection in individuals with partial humoral immunity [15] and following the development of autologous anti-NTD antibodies during acute infection in one immunocompetent individual [16]. Interestingly, NTD deletions 𝚫141 and 𝚫142 were among the selected forecasted mutations that may contribute to SARS-CoV-2 VOCs shortly, according to a recent study [17]. These findings support that P.1 variants bearing NTD deletions detected in the Amazonas and other Brazilian states [8] might represent a primary mechanism of further immune evasion of the VOC P.1.
Mutations S:P681H/R have emerged in the VOCs Alpha and Delta as well as in several VOIs (AV.1, B.1.1.318, B.1.617.1, B.1.617.3, and P.3), and reached a worldwide prevalence >70% among SARS-CoV-2 sequences sampled in May 2021 (outbreak.info SARS-CoV-2 data explorer). Mutations S:P681H/R are immediately adjacent to the S1/S2 furin cleavage site (681-P-R-R-A-R|S-686), which is a region of importance for SARS-CoV-2 transmission [18, 19], and both provide an additional basic residue adjacent to the cleavage site that may modulate S1/S2 cleavability by furin. Indeed, functional studies confirm that mutation S:P681R facilitates the furin-mediated spike cleavage and enhances viral infectivity and the efficacy of viral fusion and cell-cell viral spread, particularly when it occurs in the background of other S changes [20, 21]. Furthermore, P681R-harboring pseudoviruses were partially resistant to anti-RBD NAbs, suggesting that mutations at this position may also conformationally mask epitopes located in the RBD, blocking the accessibility of NAb to that domain [20].
Mutation S:N679K has not been previously identified as a mutation of concern. As of 25th June 2021, the EpiCoV database contains 2,585 sequences with that mutation associated with multiple SARS-CoV-2 lineages found predominantly in Europe (n =1,298) and North America (n = 1,129). The first appearance of this mutation occurred in the USA on 18th March 2020, but 97% of SARS-CoV-2 sequences with mutation S:N679K were detected from November 2020 onwards. In Europe, mutation S:N679K is found together with mutations of concern in the S protein of SARS-CoV-2 lineages B.1.1.433 (S477R), AT.1 (𝚫136-144, E484K and ins679GIAL) and B.1.258 (𝚫69-70 and N439K) and was also sporadically detected in a few B.1.1.7 (n = 30) and P.1 (n = 19) genomes.
The position of mutation S:N679K close to the furin cleavage site, the change for an additional basic residue, and its recurrent emergence on backgrounds with mutations of concern in the S protein is striking and warrants additional studies to analyze its potential functional consequences of this mutation. The furin enzyme cleavage site is composed of negatively charged residues (ASP154, ASP191, GLU236, ASP264, ASP306) that contact the substrate, forming a pocket with a remarkable negative electrostatic surface. The furin cleavage site on protein S is composed of positively charged residues so that there is a complementarity not only in shape, but also in charge between the enzyme and the substrate. Mutations N679K and P681H exchange neutral residues for two positively charged residues adjacent to the cleavage sites. We hypothesize that the positive charge increase will benefit enzyme-substrate coupling.
In summary, our study confirms that the persistent circulation of SARS-CoV-2 after the second COVID-19 epidemic wave in the Amazonas state has been associated with the continuous evolution of the VOC P.1 through the acquisition of either Spike NTD deletions or mutations at the S1/S2 junction (N679K or P681H). The rapid spread of emergent variants P.1+𝚫141-144AM, P.1+N679KAM, P.1+P681HAM, and P.1+P681HBR during May 2021 suggests that those P.1 sub-lineages are likely to continue to spread over the coming months in Amazonas and other Brazilian states. Although the emergence of these new viral variants was not associated with a third COVID-19 epidemic wave in the Amazonas state until now, the community transmission of second-generation VOCs that could be more transmissible or resistant to neutralization than the parental one should be carefully monitored. Our findings also highlight the urgent need to address the efficacy of sera from P.1-infected and vaccinated individuals to neutralize these new emergent SARS-CoV-2 P.1 sub-lineages.
References
1 Fundação de Vigilância em Saúde do Amazonas. Boletim diaria COVID-19 no Amazonas 15-06-2021, https://www.fvs.am.gov.br/media/publicacao/15_06_21_BOLETIM_DIÁRIO_DE_CASOS_COVID-19.pdf (2021).
2 Naveca, F. G. et al. COVID-19 in Amazonas, Brazil, was driven by the persistence of endemic lineages and P.1 emergence. Nat Med (2021). https://www.nature.com/articles/s41591-021-01378-7.
3 Fujino, T. et al. Novel SARS-CoV-2 Variant Identified in Travelers from Brazil to Japan. Emerg Infect Dis 27, doi:10.3201/eid2704.210138 (2021).
4 Faria, N. R. et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science, doi:10.1126/science.abh2644 (2021).
5 Konings, F. et al. SARS-CoV-2 Variants of Interest and Concern naming scheme conducive for global discourse. Nat Microbiol 6, 821-823, doi:10.1038/s41564-021-00932-w (2021).
6 Governo do Estado do Amazonas & Fundação de Vigilância em Saúde do Amazonas. Vacinação COVID-19, https://www.fvs.am.gov.br/indicadorSalaSituacao_view/75/2 (2021).
7 Governo do Estado do Amazonas & Fundação de Vigilância em Saúde do Amazonas. COVID-19 no Amazonas Dados Epidemiológicos | Boletins e Painéis de Monitoramento de Indicadores, https://www.fvs.am.gov.br/transparenciacovid19_dadosepidemiologicos (2021).
8 Resende, P. C. et al. The ongoing evolution of variants of concern and interest of SARS-CoV-2 in Brazil revealed by convergent indels in the amino (N)-terminal domain of the Spike protein. medRxiv, doi:10.1101/2021.03.19.21253946 (2021).
9 Rambaut, A., Lam, T. T., Max Carvalho, L. & Pybus, O. G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol 2, vew007, doi:10.1093/ve/vew007 (2016).
10 Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol 4, vey016, doi:10.1093/ve/vey016 (2018).
11 McCarthy, K. R. et al. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science 371, 1139-1142, doi:10.1126/science.abf6950 (2021).
12 Wang, R. et al. Analysis of SARS-CoV-2 variant mutations reveals neutralization escape mechanisms and the ability to use ACE2 receptors from additional species. Immunity, doi:10.1016/j.immuni.2021.06.003 (2021).
13 Avanzato, V. A. et al. Case Study: Prolonged Infectious SARS-CoV-2 Shedding from an Asymptomatic Immunocompromised Individual with Cancer. Cell 183, 1901-1912 e1909, doi:10.1016/j.cell.2020.10.049 (2020).
14 Chen, L. et al. Emergence of multiple SARS-CoV-2 antibody escape variants in an immunocompromised host undergoing convalescent plasma treatment. medRxiv, doi:10.1101/2021.04.08.21254791 (2021).
15 Truong, T. T. et al. Persistent SARS-CoV-2 infection and increasing viral variants in children and young adults with impaired humoral immunity. medRxiv, doi:10.1101/2021.02.27.21252099 (2021).
16 Ko, S. H. et al. High-throughput, single-copy sequencing reveals SARS-CoV-2 spike variants coincident with mounting humoral immunity during acute COVID-19. PLoS Pathog 17, e1009431, doi:10.1371/journal.ppat.1009431 (2021).
17 Maher, M. C. et al. Predicting the mutational drivers of future SARS-CoV-2 variants of concern. medRxiv, doi:10.1101/2021.06.21.21259286 (2021).
18 Johnson, B. A. et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 591, 293-299, doi:10.1038/s41586-021-03237-4 (2021).
19 Peacock, T. P. et al. The furin cleavage site of SARS-CoV-2 spike protein is a key determinant for transmission due to enhanced replication in airway cells. bioRxiv, doi:10.1101/2020.09.30.318311 (2020).
20 Saito, A. et al. SARS-CoV-2 spike P681R mutation enhances and accelerates viral fusion. bioRxiv, doi:10.1101/2021.06.17.448820 (2021).
21 Frazier, L. E. et al. Spike protein cleavage-activation mediated by the SARS-CoV-2 P681R mutation: a case-study from its first appearance in variant of interest (VOI) A.23.1 identified in Uganda. bioRxiv, doi:10.1101/2021.06.30.450632 (2021).
Data availability. All the SARS-CoV-2 genomes generated and analyzed in this study are available at EpiCoV database in GISAID (https://www.gisaid.org) with the Accession IDs showed in the Appendix Table 1.
Appendix Table1 - gisaid_hcov-19_2021_07_03_20_v2.pdf (618.9 KB)
Acknowledgements
This study was conducted as a request of the SARS-CoV-2 surveillance program of FVS-AM. It was approved by the Ethics Committee of the Amazonas State University (CAAE: 25430719.6.0000.5016), which waived the signed informed consent and the Brazilian Ministry of the Environment (MMA) SISGEN (A1767C3). The authors wish to thank all the health care workers and scientists who have worked hard to deal with this pandemic threat, the GISAID team, and all the EpiCoV database’s submitters (GISAID acknowledgment table containing sequences used in this study is attached to this post Appendix Table 2
Appendix Table 2 - gisaid_hcov-19_acknowledgement_table_2021_07_03_20.pdf (21.5 KB)
). We also appreciate the support of the Fiocruz COVID-19 Genomic Surveillance Network (http://www.genomahcov.fiocruz.br/) members, the Respiratory Viruses Genomic Surveillance. General Coordination of the Laboratory Network (CGLab), Brazilian Ministry of Health (MoH), Brazilian States Central Laboratories (LACENs), Brazilian Ministry of Health (MoH), and the Amazonas surveillance teams for the partnership in viral surveillance in Brazil. Financial support was provided by FAPEAM (PCTI-EmergeSaude/AM call 005/2020 and Rede Genômica de Vigilância em Saúde - REGESAM); Ministério da Ciência, Tecnologia, Inovações e Comunicações/Conselho Nacional de Desenvolvimento Científico e Tecnológico - CNPq/Ministério da Saúde - MS/FNDCT/SCTIE/Decit (grants 402457/2020-9 and 403276/2020-9); Inova Fiocruz/Fundação Oswaldo Cruz (Grants VPPCB-007-FIO-18-2-30 and VPPCB-005-FIO-20-2-87) and INCT-FCx (465259/2014-6). Computer allocation was partly granted by the Brazilian National Scientific Computing Center (LNCC). FGN, GLW, and GB are supported by the CNPq through their productivity research fellowships (306146/2017-7, 303902/2019-1, 425997/2018-9 and 302317/2017-1 respectively). G.B. is also funded by the Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro – FAPERJ (Grant number E-26/202.896/2018).