Emergence and spread of SARS-CoV-2 lineage B.1.1.7 in Brazil
Authors: Filipe Romero Rebello Moreira 1, Diego Menezes Bonfim 2, Danielle Alves Gomes Zauli 3, Joice do Prado Silva 3, Aline Brito de Lima 3, Frederico Scott Varella Malta 3, Alessandro Clayton de Souza Ferreira 3, Victor Cavalcanti Pardini 3, Daniel Costa Queiroz 2, Rafael Marques de Souza 2, Victor Emmanuel Viana Geddes 2, Walyson Coelho Costa 2, Wagner Carlos Santos Magalhaes 2, Rennan Garcias Moreira 4, Carolina Moreira Voloch 1, Renan Pedra de Souza 2, Renato Santana Aguiar 2,5.
1 Departamento de Genética, Instituto de Biologia, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
2 Laboratório de Biologia Integrativa, Departamento de Genética, Ecologia e Evolução, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil
3 Instituto Hermes Pardini, Belo Horizonte, Brazil
4 Centro de Laboratórios Multiusuários, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil.
5 Instituto D'Or de Pesquisa e Ensino (IDOR), Rio de Janeiro, Brazil.
Summary:
We performed a genomic surveillance study focused on the detection of lineage B.1.1.7 across eight federal states spread throughout four out of the five politically defined Brazilian regions. Our targeted approach consisted in retrospectively screening samples from the Hermes Pardini Institute, a private diagnostic company that is present in all Brazilian regions. Dataset was filtered for samples presenting S gene target failure (SGTF) with N gene amplification (Ct < 30), a signature reported in the Thermo Fisher's COVID-19 assay corresponding to the Spike 69/70 deletion. All samples were collected in January 2021 (n = 25). Sequencing was performed using the Illumina MiSeq instrument with standard ARTIC protocol. PANGOLIN confirmed all samples belonged to lineage B.1.1.7 and further phylogenetic analysis performed on a representative dataset suggests at least 21 different introductions occurred in the country. Most introductions were associated with transmission clusters comprehending samples from all evaluated states (n = 13/21; 61.9%). In addition, molecular clock analysis indicated the first introduction detected occurred in early December 2020, in agreement with the detection of lineage B.1.1.7 in São Paulo later this month. Overall, our results highlight the circulation and spread of this variant of concern in Brazil, in extension to previous reports of lineages P.1 and P.2.
Context:
SARS-CoV-2 was introduced in Brazil in March 2020 and caused massive epidemic waves throughout the country (1). These introduction events were followed by the emergence of several lineages, which led to continuous chains of transmissions that culminated in more than 10 million cases and 250,000 deaths reported in Brazil to the present time (https://coronavirus.jhu.edu/map.html). More recently, in several regions of the world, variants of concern (VOCs) have been detected and associated with a rise in the number of cases due to their increased infectivity. Examples include lineages B.1.1.7 (2) and B.1.1.351 (3) in the UK and South-Africa, respectively. In Brazil, two lineages have been reported almost simultaneously in Amazonas (P.1 (4)) and Rio de Janeiro (P.2 (5)) states, sharing mutations with B.1.1.7 and B.1.1.351 putatively associated with increasing transmissibility of COVID-19.
Study:
Given the epidemiological importance of lineage B.1.1.7 and reports of its circulation in São Paulo state since December, 2020 (6), we were prompted to investigate its circulation and spread in Brazilian territory. Samples were obtained from Hermes Pardini Institute, one of the largest Brazilian diagnostic companies averaging 240,000 COVID-19 tests per month across all Brazilian states. Among other mutations, B.1.1.7 carries the Spike 69/70 deletion leading to the S gene target failure (SGTF; 7) reported in the Thermo Fisher's COVID-19 assay that has being used by Hermes Pardini laboratory in COVID-19 diagnose since May 2020. Therefore, we retrospectively filtered our dataset for positive samples presenting N gene amplification (Ct < 30) and SGTF. We obtained 25 samples collected between January 4th and 24th 2021 that met our criteria in eight different states scattered along four out of the five Brazilian geopolitical regions: Northeast (Bahia, Sergipe), Central-West (Mato Grosso), Southeast (Espírito Santo, Minas Gerais, Rio de Janeiro and São Paulo) and South (Paraná). Amplified fragments spanning the whole genome of SARS-CoV-2 and DNA libraries were prepared using QIAseq SARS-CoV-2 Primer Panel and QIAseq FX DNA Library Kit, respectively. Sequencing was performed in the Illumina MiSeq instrument with a V3-600 Illumina cartridge. Samples metadata and sequencing statistics are available in table 1.
Table 1: Samples metadata and basic sequencing statistics.
| Sample | Accession | Collection date | Location(state,municipality) | Ct N | Genome coverage 10x (%) | Average depth |
|---|---|---|---|---|---|---|
| SEQ10 | EPI_ISL_1133256 | 2021-01-07 | Paraná, Curitiba | 15.46 | 99.58 | 1462.13 |
| SEQ11 | EPI_ISL_1133257 | 2021-01-07 | Minas Gerais, Belo Horizonte | 18.64 | 89.02 | 232.18 |
| SEQ1B | EPI_ISL_1133271 | 2021-01-12 | Minas Gerais, Belo Horizonte | 12.5 | 81.05 | 51.70 |
| SEQ02 | EPI_ISL_1133272 | 2021-01-13 | Minas Gerais, Belo Horizonte | 14.5 | 98.91 | 1442.43 |
| SEQ25 | EPI_ISL_1133279 | 2021-01-24 | Mato Grosso, Cuiabá | 15.31 | 87.53 | 42.10 |
| SEQ28 | EPI_ISL_1133262 | 2021-01-09 | Minas Gerais, Belo Horizonte | 17.84 | 98.46 | 3148.76 |
| SEQ29 | EPI_ISL_1133263 | 2021-01-09 | Minas Gerais, Belo Horizonte | 14.2 | 99.76 | 2405.53 |
| SEQ03 | EPI_ISL_1133273 | 2021-01-14 | Minas Gerais, Belo Horizonte | 16.2 | 98.98 | 1414.17 |
| SEQ30 | EPI_ISL_1133265 | 2021-01-11 | Minas Gerais, Belo Horizonte | 15.49 | 99.59 | 5432.8 |
| SEQ31 | EPI_ISL_1133269 | 2021-01-12 | Minas Gerais, Belo Horizonte | 16.15 | 98.92 | 4030.44 |
| SEQ33 | EPI_ISL_1133264 | 2021-01-11 | Rio de Janeiro, Rio de Janeiro | 17.91 | 96.79 | 190.01 |
| SEQ34 | EPI_ISL_1133258 | 2021-01-08 | São Paulo, Americana | 18.85 | 99.03 | 2846.06 |
| SEQ36 | EPI_ISL_1133266 | 2021-01-11 | Rio de Janeiro, Campos dos Goytacazes | 16.65 | 99.76 | 3884.28 |
| SEQ37 | EPI_ISL_1133259 | 2021-01-08 | São Paulo, Santos | 13.53 | 99.09 | 2450.99 |
| SEQ38 | EPI_ISL_1133277 | 2021-01-18 | Bahia, São Sebastião do Passe | 18.45 | 99.67 | 2790.42 |
| SEQ39 | EPI_ISL_1133274 | 2021-01-14 | Mato Grosso, Primavera do Leste | 21.44 | 99.65 | 3325.17 |
| SEQ04 | EPI_ISL_1133270 | 2021-01-12 | São Paulo, Valinhos | 21.5 | 93.87 | 751.50 |
| SEQ41 | EPI_ISL_1133278 | 2021-01-21 | Minas Gerais, Betim | 14.44 | 99.66 | 3500.7 |
| SEQ43 | EPI_ISL_1133276 | 2021-01-15 | Minas Gerais, Belo Horizonte | 15.13 | 98.84 | 3033.23 |
| SEQ44 | EPI_ISL_1133267 | 2021-01-12 | Espírito Santos, Barra do São Francisco | 13.97 | 99.77 | 2317.93 |
| SEQ45 | EPI_ISL_1133268 | 2021-01-12 | Rio de Janeiro, Rio de Janeiro | 16.0 | 99.15 | 2737.82 |
| SEQ48 | EPI_ISL_1133275 | 2021-01-15 | Minas Gerais, Barbacena | 23.24 | 98.79 | 3716.37 |
| SEQ07 | EPI_ISL_1133255 | 2021-01-04 | Minas Gerais, Araxá | 14.73 | 96.74 | 1612.66 |
| SEQ09 | EPI_ISL_1133260 | 2021-01-08 | Minas Gerais, Belo Horizonte | 16.95 | 98.35 | 889.91 |
| SEQ35 | EPI_ISL_1133261 | 2021-01-08 | Sergipe, Aracajú | 15.67 | 99.76 | 3377.61 |
Quality control of the obtained sequencing reads was performed with Trimmomatic v0.39 (8). Reads were then mapped against the SARS-CoV-2 reference genome (NC_045512.2) with Bowtie2 (9). SAMtools (10), VCFtools (11) and BEDtools (12) were used to infer consensus genome sequences. All sequenced samples had genome coverage above 75% (mean 10x coverage of 97.3%, range: 79.8-99.8%; mean 100x coverage of 88%, range: 5-98.8%).
All consensus genome sequences were screened with PANGOLIN software (13), which confirmed all samples belonged to lineage B.1.1.7, consistent with our pre-screening strategy. A representative dataset of this lineage was then assembled from sequences available on GISAID, containing one international sequence per week per country since the discovery of this lineage until February 18th 2021. All 22 previously described Brazilian B.1.1.7 sequences (one from Goiás, two from Distrito Federal and 19 from São Paulo) in GISAID and the new genomes generated here were also included. A maximum likelihood tree was then inferred on IQ-tree 2 (14) under the GTR+F+I model (15), suggested by its built in model selection algorithm. This phylogenetic tree supports the occurrence of 13 Brazilian clades containing between two and five sequences from diverse states, suggesting multiple introductions occurred in the country, leading to local transmission chains (Figure 1). A clade was termed as Brazilian if at least half its sequences belonged to the country. Noticeably, while some clades were clearly related to specific states, as São Paulo or Minas Gerais, others contained sequences from up to three states, revealing the connection among locations as a driver of lineage interiorization. In addition, eight unique sequences grouped separately with international samples, suggesting up to 21 introductions occurred (even though this is very likely an underestimate, given the low number of samples evaluated).
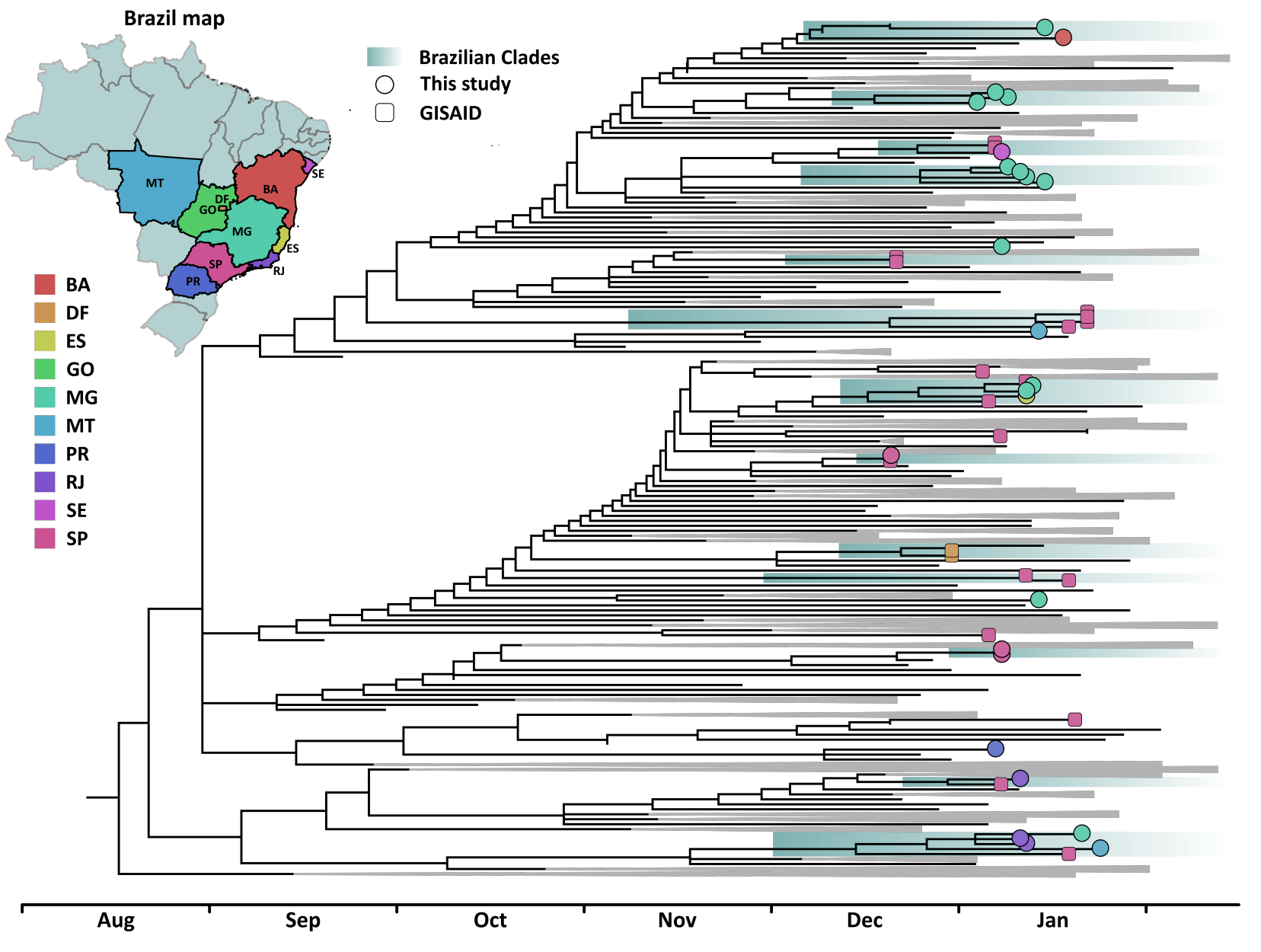
Figure 1: Time scaled phylogenetic tree inferred from a dataset comprehending 406 publicly available B.1.1.7 sequences and the 25 new genomes characterized in this study. Brazilian sequences are color coded according to originary federal states and tip shapes mark new and previously described genome sequences. Brazilian clades are shaded in green. The tree supports that multiple introductions occurred in different regions of the country between early December 2020 and early January 2021. While some introductions are related to single sequences, others are linked with the emergence of clades, emphasizing the occurrency of local transmission in the country.
The imbalance between sampled locations in the dataset precluded the use of standard discrete phylogeographic models, though we think it is worth to notice that Brazilian sequences clustered with sequences from several countries from Africa, Asia and Europe, reinforcing the importance of genomic surveillance and increasing numbers of SARS-CoV-2 sequences from Brazil. In regards to dating these introductions, a molecular clock analysis was performed with TreeTime (16), using a fixed previously estimated evolutionary rate (1). The Brazilian clades have been dated between early December 2020 and early January 2021 (oldest clade: December 8th 2020, 95% CI: 3 - 9 December 2020; youngest clade: January 12th, 2021, 95% CI: December 22th 2020 - January 12th 2021). These dates are consistent with an early report on the detection of lineage B.1.1.7 in São Paulo state (6). Notwithstanding, SGTF first detection in Hermes Pardini Institute was on October 16th 2020 in São José do Rio Preto, São Paulo state. This observation could imply that this lineage has been circulating even before than the dates herein estimated, a conjecture that further sequencing should confirm.
Overall, our results support that a targeted approach using SGTF was effective to track and identify samples infected by SARS-CoV-2 lineage B.1.1.7, as previously reported (7). Phylogenetic analysis performed with genome sequences obtained from these samples showed that this lineage was introduced multiple times in different regions of Brazil, leading to continuous chains of transmissions since early December 2020. This lineage has now been identified in ten different states in nearly all regions of the country and the consequences of this spread remains unknown. Whether its circulation will impact the Brazilian epidemiological scenario, as occurred in the UK, is yet to be revealed. Nevertheless, it also remains unknown if the co-circulation of lineage B.1.1.7 with other VOCs (P.1 and P.2) will lead to increased transmissibility. Future assessments should focus on identifying the repercussion of these lineages circulation on epidemic curves and seek functional explanations for these effects.
Limitations:
Our study has been performed with a limited set of samples (n = 25) restricted to January 2021. Even though these samples have been collected from several different states, confirming circulation of lineage B.1.1.7 across the country, it is almost certain that the number of introductions inferred is underestimated. Likewise, it is possible that further sequencing of samples from previous dates might reveal introduction dates earlier than the ones estimated here. Our study was also not able to fully explore the epidemiological consequences of the circulation of this lineage in the country, nor the consequence of co-circulation with other Brazilian variants of concern (P.1 and P.2), matters that follow-up studies should address.
Data sharing:
Genome sequences generated in this study have been deposited on GISAID (Accessions: EPI_ISL_1133259, EPI_ISL_1133273, EPI_ISL_1133274, EPI_ISL_1133275, EPI_ISL_1133276, EPI_ISL_1133255, EPI_ISL_1133277, EPI_ISL_1133256, EPI_ISL_1133278, EPI_ISL_1133257, EPI_ISL_1133279, EPI_ISL_1133258, EPI_ISL_1133270, EPI_ISL_1133271, EPI_ISL_1133272, EPI_ISL_1133262, EPI_ISL_1133263, EPI_ISL_1133264, EPI_ISL_1133265, EPI_ISL_1133266, EPI_ISL_1133267, EPI_ISL_1133268, EPI_ISL_1133269, EPI_ISL_1133260, EPI_ISL_1133261).
Acknowledgments:
We would like to thank all authors who submitted their data to GISAID, allowing this genomic epidemiology study to be properly conducted. A full list of acknowledgment is available in table 2.
Funding:
We acknowledge support from the Rede Corona-ômica BR MCTI/FINEP affiliated to RedeVírus/MCTI (FINEP 01.20.0029.000462/20, CNPq 404096/2020-4). This project was also supported by CNPq (R.S.A.: 312688/2017-2 and 439119/2018-9; R.P.S.: 310627/2018-4), MEC/CAPES (14/2020 - 23072.211119/2020-10), FINEP (0494/20 01.20.0026.00), FAPEMIG (R.P.S.: APQ-00475-20) and FAPERJ (C.M.V: 26/010.002278/2019).
References:
-
Candido, D. S. et al. Evolution and epidemic spread of SARS-CoV-2 in Brazil. Science 1260, 1255–1260 (2020).
-
Volz, E. et al. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. medRxiv 2020.12.30.20249034 (2021).
-
Tegally, H. et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv 2, (2020).
-
Voloch, C. M. et al. Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro, Brazil. medRxiv (2020).
-
Claro, I. M. et al. Local Transmission of SARS-CoV-2 Lineage B.1.1.7, Brazil, December 2020. 27 Emerging Infectious Diseases, 2020–2022 (2021).
-
Bal, A. et al. Two-step strategy for the identification of SARS-CoV-2 variant of concern 202012/01 and other variants with spike deletion H69-V70, France, August to December 2020. medRxiv 33, (2021).
-
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
-
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
-
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
-
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
-
Quinlan, A. R. & Hall, I. M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
-
Minh, B. Q. et al. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 37, 1530–1534 (2020).
-
Tavaré, S. Some probabilistic and statistical problems in the analysis of DNA sequences. American Mathematical Society: Lectures on Mathematics in the Life Sciences vol. 17 57–86 (1986).
-
Sagulenko, P., Puller, V. & Neher, R. A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 4, 1–9 (2018).