Natalia Rego1*, Cecilia Salazar2*, Mercedes Paz3*, Alicia Costábile3,4,5*, Alvaro Fajardo4,5, Ignacio Ferrés2, Paula Perbolianachis4,5, Tamara Fernández-Calero1, Veronica Noya6, Rodrigo Arce4,5, Mailen Arleo6, Tania Possi6, Natalia Reyes6, María Noel Bentancor6, Andrés Lizasoain7, Viviana Bortagaray7, Ana Moller7, Odhille Chappos8, Nicolas Nin9, Javier Hurtado9, Melissa Duquía8, Belén González8, Luciana Griffero8, Mauricio Méndez8, Ma Pía Techera8, Juan Zanetti8, Bernardina Rivera10, Matías Maidana10, Martina Alonso10, Cecilia Alonso8, Julio Medina11, Henry Albornoz11, Rodney Colina7, Gonzalo Bello12, Pilar Moreno4,5#, Gonzalo Moratorio4,5#, Gregorio Iraola2#, Lucía Spangenberg1#
1 Bioinformatics Unit, Institut Pasteur de Montevideo, Uruguay.
2 Laboratorio de Genómica Microbiana, Institut Pasteur Montevideo, Uruguay.
3 Centro de Innovación en Vigilancia Epidemiológica, Institut Pasteur Montevideo, Uruguay.
4 Laboratorio de Virología Molecular, Facultad de Ciencias, Universidad de la República, Uruguay.
5 Laboratorio de Evolución Experimental de Virus, Institut Pasteur de Montevide, Uruguay.
6 Laboratorio de Biología Molecular, Sanatorio Americano, Uruguay
7 CENUR Litoral Norte, Universidad de la República, Salto, Uruguay
8 Centro Universitario Regional Este, Universidad de la República, Rocha, Uruguay.
9 Hospital Español, Uruguay
10 Laboratorio de Diagnóstico Molecular, Institut Pasteur de Montevideo, Uruguay
11 Facultad de Medicina, Universidad de la República, Uruguay
12 Laboratorio de AIDS e Imunologia Molecular, Instituto Oswaldo Cruz, Fiocruz, Rio de Janeiro, Brazil.
Summary
Uruguay was able to control the early viral dissemination during the first nine months of the SARS-CoV-2 pandemic. Unfortunately, towards the end of 2020, the number of daily new cases exponentially increased which coincided with the loss of the TETRIS safety zone. In a previous study we found a sublineage B.1.1.28+Q675H+Q677H with local transmission in Rocha, a Uruguayan department bordering Brazil. This clade probably arose by early November, 2020, and its introduction from other parts of Uruguay seemed like a reasonable hypothesis. To understand whether these sequences were part of a new emergent SARS-CoV-2 lineage broadly disseminated in Uruguay, herein we analyzed the genetic diversity of B.1.1.28 SARS-CoV-2 viruses circulating in different localities by the end of 2020 and first months of 2021. The spatiotemporal reconstruction using 89 new sequences sampled from December 2020 to April 2021, further supports that this B.1.1.28+Q675H+Q677H clade probably arose around November 2020, in Montevideo, the capital department of Uruguay. This clade further spread from Montevideo to other Uruguayan departments, with evidence of local transmission clusters in Rocha, Salto and Tacuarembo. It also spread to the USA and Spain. The non-synonymous mutations in the viral Spike Q675H and Q677H, which are among the 10 lineage-defining mutations, are in **the proximity of the polybasic cleavage site at the S1/S2 boundary and also are reported as recurrent arising independently in many SARS-CoV-2 lineages circulating worldwide by the end of 2020. Although the B.1.1.28+Q675H+Q677H lineage was later substituted by the VOC P.1 as the most prevalent lineage in Uruguay since April 2021, the monitoring of the concurrent emergence of Spike mutations Q675H+Q677H in VOIs and/or VOCs should be of worldwide interest.
Background
Uruguay was able to control the early viral dissemination during the first nine months of the SARS-CoV-2 pandemic. Unfortunately, towards the end of 2020, the number of new cases exponentially increased, from 60 cases per day on average during October and November to over 400 during December. This coincided with the loss of the TETRIS (Test, Trace and Isolation strategy) safety zone [1,2]. With a 1,068 km long Uruguayan-Brazilian dry border, multiple introductions and successful dissemination of Brazilian lineages B.1.1.28, B.1.1.33, P.1 and P.2 have occurred during 2020 and 2021 [3-5]. Little is known regarding the factors related to SARS-CoV-2 viral dynamics during the first exponential increase of COVID-19 cases by the end of 2020, before the arrival of VOI P.2 and VOC P.1 (which earliest samples are from January and February 2021, respectively).
In a previous study we surveyed patients diagnosed between November 2020 and February 2021 in Rocha, an eastern Uruguayan department bordering Brazil, we found that lineage B.1.1.28 was the most prevalent during November-December, 2020 [5]. Many B.1.1.28 sequences branched in a clade highly prevalent in southern Brazil, while others branched in a second monophyletic cluster harboring several lineage-defining mutations including two non-synonymous changes in the Spike protein: Q675H and G677H, so far not concurrently reported. The convergent appearance of S:Q677H in different viral lineages and its proximity to the S1/S2 cleavage site raised concerns about its functional relevance [6]. The spatiotemporal reconstruction suggested that this clade probably arose by early November, 2020, with a viral introduction into the city of Rocha (capital of the homonymous department), but a prior dissemination of this sublineage in other parts of Uruguay before being introduced in Rocha seemed like a reasonable assumption. To understand whether these sequences were part of a new emergent SARS-CoV-2 lineage broadly disseminated in Uruguay, herein we analyzed the genetic diversity of B.1.1.28 SARS-CoV-2 viruses circulating in different localities by the end of 2020 and first months of 2021.
The Study
The Uruguayan inter-institutional working group (IiWG) for SARS-CoV-2 genomic surveillance sequenced 113 B.1.1.28 SARS-CoV-2 positive samples (Appendix 1) detected in Uruguay between August 7, 2020 and May 30, 2021, with procedures previously explained in [4]. The mutational profile identified 99 sequences (88%) carrying both S:Q675H and S:Q677H amino acid changes (Figure 1A). The B.1.1.28+Q675H+Q677H sequences were widely spread throughout the country, being detected in 12 out of 19 Uruguayan departments from 2nd December, 2020 to 25th April, 2021.
All B.1.1.28 Uruguayan sequences here obtained were combined with B.1.1.28 complete genome sequences available at the EpiCoV database in GISAID by June 07, 2021 sampled in Uruguay (n=184) and Brazil (n=1,428) and with all B.1.1.28 sequences sampled worldwide (USA=2, Spain=1 and Belgium=1) that carry mutations Q675H and Q677H (B.1.1.28-UY/GISAID_hcov-19_2021_07_07_20_TS1.csv at main · natinreg/B.1.1.28-UY · GitHub). A Maximum-likelihood (ML) phylogenetic tree was reconstructed using IQ-TREE v2.1.2 [7], the time-scale was inferred using TreeTime [8] applying a fixed clock rate of 8 x 10 -4 substitutions/site/year, based on previous estimates [9], and then implemented the ancestral character state reconstruction (ACR) of epidemic locations available in PastML [10], using the Marginal Posterior Probabilities Approximation (MPPA) method with an F81-like model. This analysis revealed that all B.1.1.28+Q675H+Q677H sequences from Uruguay (n=246) branched in a highly supported (approximate likelihood-ratio test [aLRT] = 99.5%) monophyletic clade designated UY-I (Figure 1B). According to the ML-based ACR of epidemic locations, the UY-I clade was most likely introduced from the southeastern Brazilian region (ACR location marginal probability = 0.99) and was disseminated from Uruguay to the USA and Spain.
We next conducted a Bayesian phylogeographic reconstruction using BEAST 1.10 [11] for the UY-I sequences generated in this study (n=110) plus six additional B.1.1.28 basal sequences from Brazil. The inference used the GTR+F+I nucleotide substitution model, the nonparametric Bayesian skyline model as the coalescent tree prior [12], a strict molecular clock model with a uniform substitution rate prior (8-10 × 10-4 substitutions/site/year) and a reversible discrete phylogeographic model [13] with a continuous-time Markov chain (CTMC) rate reference prior [14]. The spatiotemporal reconstruction suggests that this clade of UY-I probably arose around November 17, 2020 (HPD: October 29, 2020 to November 29, 2020) in Montevideo, the capital department of Uruguay (Figure 1C). This clade further spread from Montevideo to other Uruguayan departments, with evidence of local transmission clusters in Rocha, Salto and Tacuarembo. The TMRCA of local transmission clusters in Rocha and Salto was traced to December 24, 2020 (HPD: December 14, 2020 to December 31, 2020) and January 24, 2021 (HPD: December 27, 2020 to February 13, 2021), respectively (Figure 1C). The introduction and dispersion of this lineage in each department coincided with the increase in COVID-19 reported daily new cases (Figure 1D).
This new B.1.1.28 sublineage spreading in Uruguay is characterized by 10 lineage-defining genetic changes, including five non-synonymous mutations (Figure 1C). Three were located in the Spike protein being two the non-synonymous mutations Q675H and Q677H, which are in the proximity of the polybasic cleavage site at the S1/S2 boundary. The amino acid change S:Q677H has been reported as a recurrent mutation arising independently in many SARS-CoV-2 lineages circulating worldwide by the end of 2020 [6]. In fact, a search in the EpiCoV database (accessed on July 7, 2021) reported 146 samples with high quality SARS-CoV-2 genome sequences with both Q675H and Q677H present, distributed in 12 different countries (in descendent frequency order: Uruguay, England, USA, Belgium, India, Australia, Switzerland, Spain, Netherlands, Japan, Germany and France) and genotyped as 13 different pango lineages (in descendent frequency order: B.1.36, B.1.1.28, B.1.2, C.36, B.1.538, B.1.1.316, B.1.526, B.1.525, B.1.243, B.1.1.70, B.1.1.7, B.1.1.63 and B.1). Recently, a study forecasted that these mutations could appear in emerging SARS-CoV-2 VOCs in the following months [15].
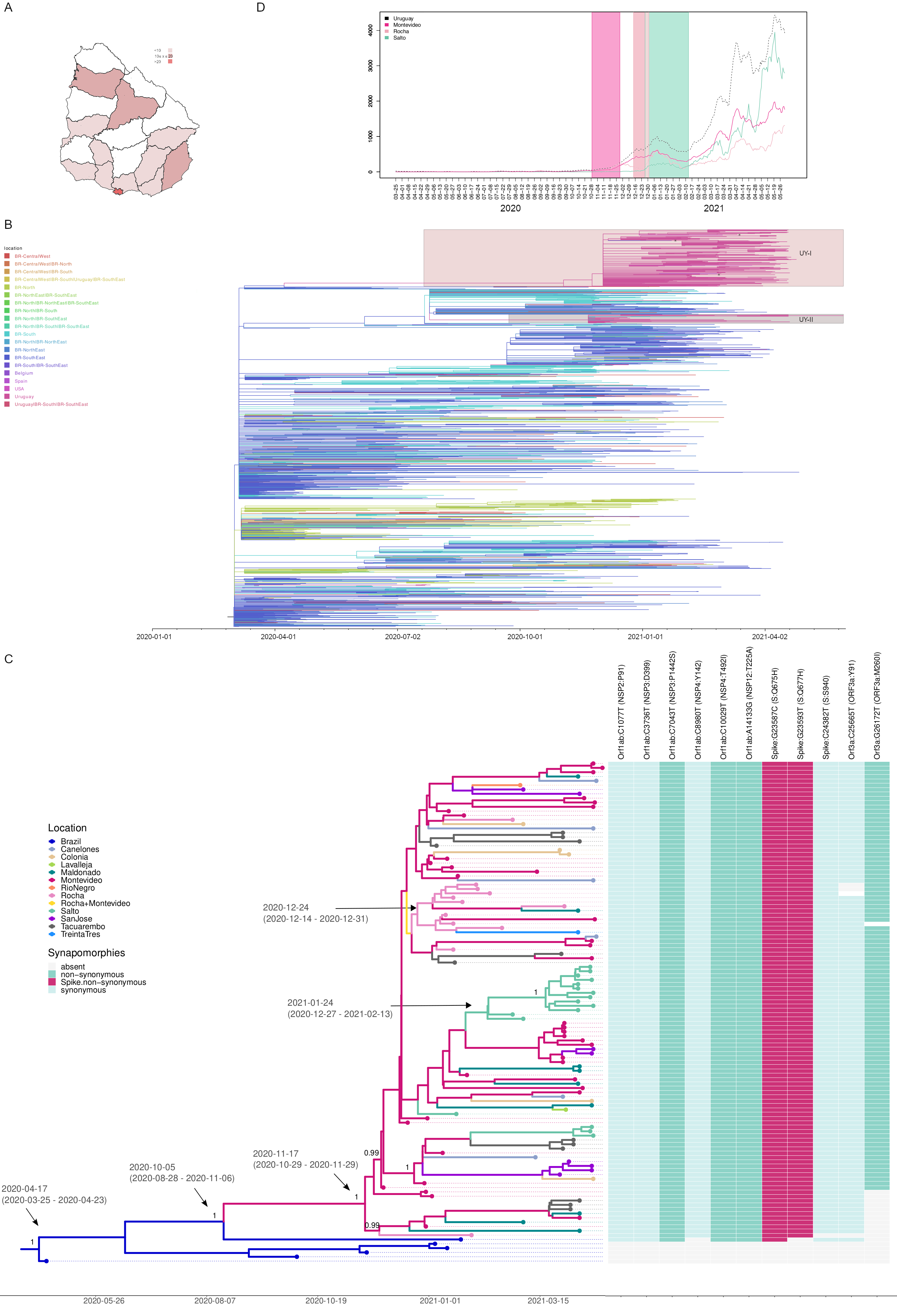
Figure 1. A Map of Uruguay showing the number of sequences classified as B.1.1.28+Q675H+Q677H in every department. B. Maximum likelihood phylogeographic analysis of lineage B.1.1.28 samples (n=1719) from Uruguay (n=246), Brazil (n=1469) and USA, Spain and Belgium (n=4) inferred by ancestral character reconstruction method implemented in PastML. Tips and branches are colored according to sampling location and the most probable location state of their descendent nodes, respectively, as indicated in the legend. Shaded boxes highlight the major B.1.1.28 clades in Uruguay. UY-I corresponds to the clade B.1.1.28+Q675H+Q677H here discussed while UY-II is a B.1.1.28 clade carrying mutation N:P13L widely distributed in southern Brazil. Asterisks indicate the sequences dispersed from Uruguay to the USA and Spain. The time scaled tree was rooted with the earliest sequence. C. Bayesian phylogeographic analysis of B.1.1.28+Q675H+Q677H clade in Uruguay, implemented in BEAST. Uruguayan sequences generated by the IiWG (n=110) were combined with six additional basal sequences from southeastern Brazil. Tips and branches of the time-scaled Bayesian tree are colored according to sampling location and the most probable location state of their descendent nodes, respectively, as indicated in the legend. Posterior probability support values and estimated TMRCAs are indicated at key nodes. Additionally, a heatmap represents the presence or absence of synapomorphic sites. The color scheme indicates the different mutations, as indicated in the legend. In each case, genomic position, nucleotide substitution, viral protein and amino acid is shown. D. Number of daily new cases from March, 2020 to May, 2021 in the country (black), in Montevideo (fuchsia), Rocha (pink) and in Salto (gren). Daily new cases for Rocha and Salto were multiplied by a factor proportional to the population of that department in comparison to Montevideo (times 23 and 13, for Rocha and Salto, respectively) for visualization purposes. Confidence intervals of TMRCA of Montevideo (fuchsia), Rocha (pink) and Salto (green) clades are shown as shaded areas.
Conclusions
This study describes the emergence and local spread of a B.1.1.28 sublineage carrying Spike mutations Q675H+Q677H in Uruguay that coincided with the first exponential growth phase of the COVID-19 epidemic in the country that started by November, 2020. The ancestral virus was probably introduced from southeastern Brazil into Montevideo, Uruguay by November 2020 and this virus rapidly disseminated across the whole country. Although the B.1.1.28+Q675H+Q677H lineage was later substituted by the VOC P.1 as the most prevalent lineage in Uruguay since April 2021, the monitoring of the concurrent emergence of Spike mutations Q675H+Q677H in VOIs and/or VOCs should be of worldwide interest.
Acknowledgments
The National Ministry of Health (Uruguay) is the main health Institution in our country. It is a dedicated ethics oversight body and has granted us the ethical approval for this work. All necessary patient/participant consent has been obtained and the appropriate institutional forms have been archived.
The authors wish to thank all the health care workers and scientists who have worked hard to deal with this pandemic threat, the GISAID team, and all the EpiCoV database’s submitters, GISAID acknowledgment table containing sequences used in this study is attached to this post (Appendix 2). This work was supported by FOCEM-Fondo para la Convergencia Estructural del Mercosur (COF03/11).
Data availability
All SARS-CoV-2 genome sequences have been submitted to the EpiCoV/GISAID database with accession numbers indicated in the linked table.
References
[1] C Fraser, S Riley, RM Anderson, NM Ferguson. Factors that make an infectious disease outbreak controllable. Proc Natl Acad Sci U S A. 2004, 101(16):6146-51. doi:10.1073/pnas.0307506101.
[2] KH Grantz, EC Lee, ML D’Agostino, KH Lee, CJE Metcalf, et al. Maximizing and evaluating the impact of test-trace-isolate programs: A modeling study. PLoS Med. 2021, 18(4):e1003585. doi:10.1371/journal.pmed.1003585.
[3] D Mir, N Rego, PC Resende, F Tort, T Fernández-Calero, et al. Recurrent Dissemination of SARS-CoV-2 Through the Uruguayan-Brazilian Border. Front Microbiol. 2021 May 28;12:653986. doi:10.3389/fmicb.2021.653986.
[4] N Rego, A Costábile, M Paz, C Salazar, P Perbolianachis, et al. Implementation of a qPCR assay coupled with genomic surveillance for real-time monitoring of SARS-CoV-2 variants of concern. medRxiv, 2021, doi:10.1101/2021.05.20.21256969
[5] N Rego, T Fernández-Calero, I Arantes, V Noya, D Mir, et al. Spatiotemporal dissemination pattern of SARS-CoV-2 B.1.1.28-derived lineages introduced into Uruguay across its southeastern border with Brazil. medRxiv, 2021, doi:10.1101/2021.07.05.21259760
[6] EB Hodcroft, DBDomman, DJ Snyder, KY Oguntuyo, M Van Diest, et al. Emergence in late 2020 of multiple lineages of SARS-CoV-2 Spike protein variants affecting amino acid position 677. medRxiv, 2021, doi:10.1101/2021.02.12.21251658
[7] LT Nguyen, HA Schmidt, A von Haeseler, BQ Minh. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015 Jan;32(1):268-74. doi: 10.1093/molbev/msu300. Epub 2014 Nov 3. PMID: 25371430; PMCID: PMC4271533.
[8] P Sagulenko, V Puller, RA Neher. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018 Jan 8;4(1):vex042. doi: 10.1093/ve/vex042. PMID: 29340210; PMCID: PMC5758920.
[9] S Duchene, L Featherstone, M Haritopoulou-Sinanidou, A Rambaut, P Lemey, et al. Temporal signal and the phylodynamic threshold of SARS-CoV-2. Virus Evol. 2020 Aug 19;6(2):veaa061. doi:10.1093/ve/veaa061. PMID: 33235813; PMCID: PMC7454936.
[10] SA Ishikawa, A Zhukova, W Iwasaki, O Gascuel. A Fast Likelihood Method to Reconstruct and Visualize Ancestral Scenarios. Mol Biol Evol. 2019 Sep 1;36(9):2069-2085. doi:10.1093/molbev/msz131. PMID: 31127303; PMCID: PMC6735705.
[11] MA Suchard, P Lemey, G Baele, DL Ayres, AJ Drummond et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018 Jun 8;4(1):vey016. doi:10.1093/ve/vey016. PMID: 29942656; PMCID: PMC6007674.
[12] AJ Drummond, A Rambaut, B Shapiro, OG Pybus. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol. 2005 May;22(5):1185-92. doi:10.1093/molbev/msi103. Epub 2005 Feb 9. PMID: 15703244.
[13] P Lemey, A Rambaut, AJ Drummond, MA Suchard. Bayesian phylogeography finds its roots. PLoS Comput Biol. 2009 Sep;5(9):e1000520. doi:10.1371/journal.pcbi.1000520. Epub 2009 Sep 25. PMID: 19779555; PMCID: PMC2740835.
[14] MAR Ferreira, MA Suchard. Bayesian analysis of elapsed times in continuous-time Markov chains. The Canadian Journal of Statistics 2010. doi: 10.1002/cjs.5550360302
[15] MC Maher, I Bartha, S Weaver, J di Iulio, E Ferri, et al. Predicting the mutational drivers of future SARS-CoV-2 variants of concern. medRxiv 2021, doi:10.1101/2021.06.21.21259286