On 1 June 2020, the Democratic Republic of the Congo (DRC) announced a new cluster of Ebola virus disease (EVD) cases occurring in Mbandaka health zone, in Équateur Province. This is the same area where the DRC’s 9th Ebola outbreak occurred back in May 2018. A sample was sent to Institut National de Recherche Biomédicale (INRB) in Kinshasa for confirmatory testing. Ebola virus (EBOV) was confirmed by PCR on 1 June 2020 using the GeneXpert (GP ct 31, NP ct 28). The sample was sequenced using an amplicon strategy developed with Primal Scheme (using MK007330 as a reference to generate the primers), followed by Illumina Nextera DNA Flex library preparation. The consensus genome was generated using iVar on 4 June 2020.
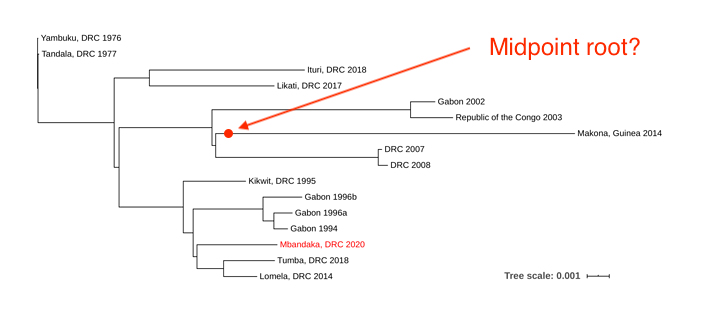
The new EVD cluster could have been seeded from the current Nord Kivu/Ituri outbreak (Ituri), from the May 2018 Équateur province outbreak (Tumba) via a persistent source, or a new spillover event from the reservoir host. The new EBOV genome was aligned to single representatives from previous EVD outbreaks using MAFFT, and a phylogenetic tree was inferred using RaxML (Figure 1).
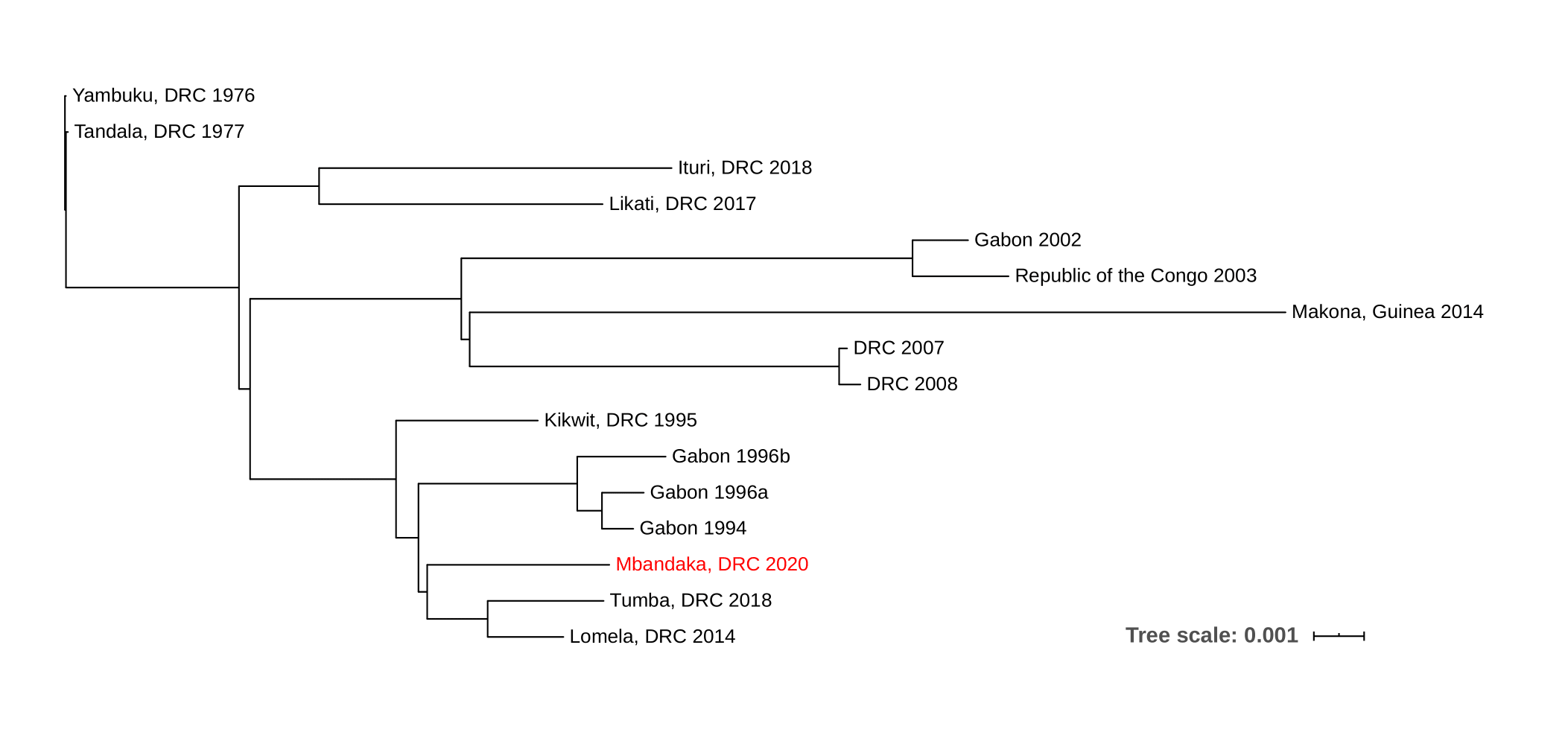
Figure 1. Maximum likelihood phylogenetic tree of all EVD outbreaks, including the sequence from the new outbreak in red.
Due to the level of divergence from Ituri (>350 substitutions) and Tumba (>150 substitutions), this genome likely represents a new spillover event and the 11th EVD outbreak in the DRC, the third outbreak in two years. A root-to-tip analysis (Figure 2) shows the new genome grouping with four variants that have reduced rates of evolution compared with the 11 other representative sequences. More thorough analyses will be conducted in the coming days.
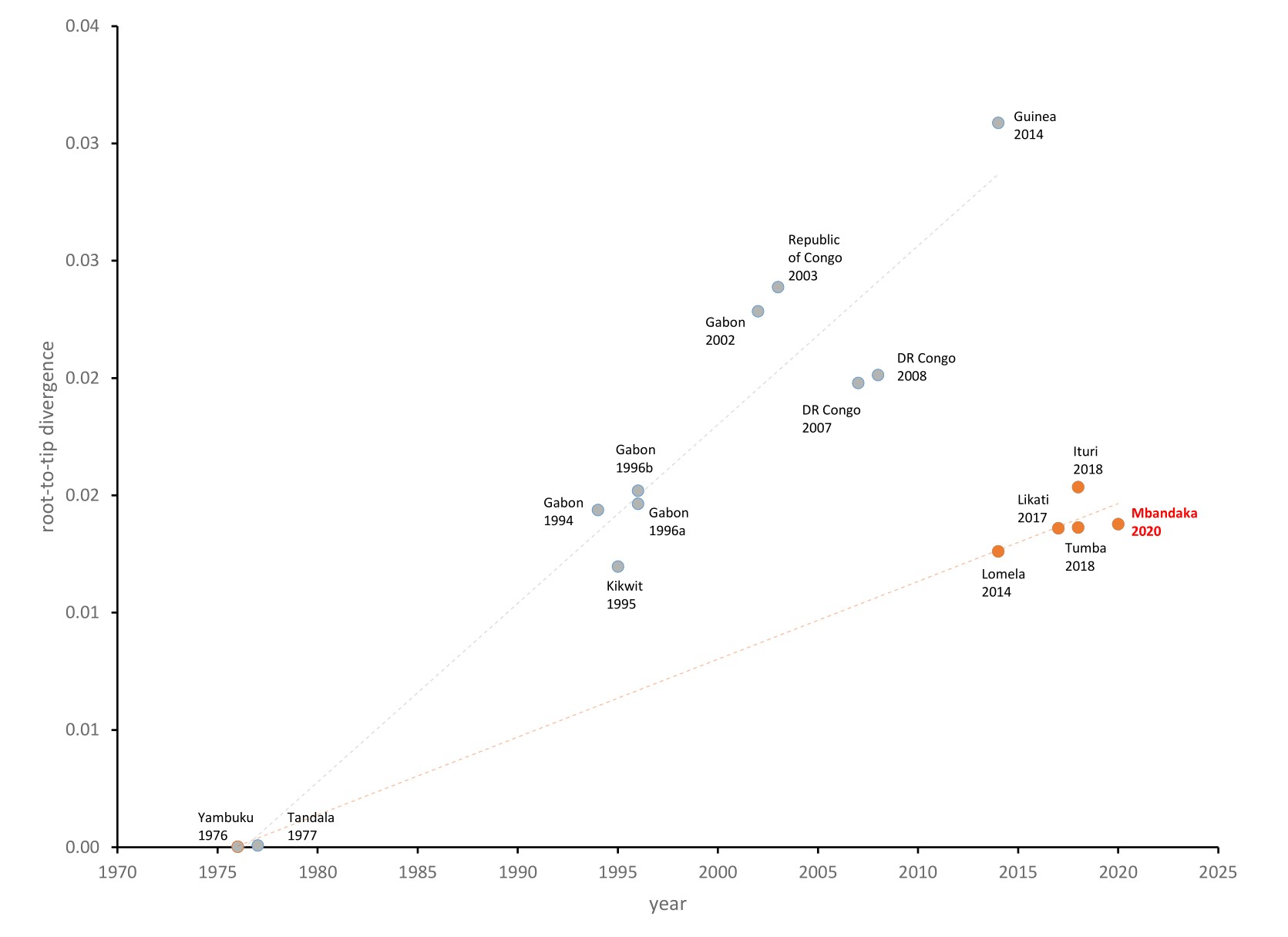
Figure 2. Inter-outbreak root-to-tip analysis. EBOV genomes from 1976 to 2014 (West Africa) (grey) and from 2014 (DRC) to 2020 (red) with the new Mbandaka genome in red text.
Partners and Collaborators
Prof. Jean-Jacques Muyembe-Tamfum
Institut National de Recherche Biomédicale (INRB); School of Medicine Kinshasa University
Prof. Steve Ahuka-Mundeke
INRB; School of Medicine Kinshasa University
Prof. Placide Mbala-Kingebeni
INRB; TransVIHMI (IRD, INSERM, University of Montpellier); School of Medicine Kinshasa University
Eddy Kinganda-Lusamaki
INRB; School of Medicine Kinshasa University
Adrienne Amuri-Aziza
INRB
Dr. Michael Wiley
University of Nebraska Medical Center
Catherine Pratt
University of Nebraska Medical Center
Statement on continuing work and analyses prior to publication
This genome is being shared pre-publication. Please note that this data is based on work in progress and should be considered preliminary. Our analyses of this data is ongoing and a publication communicating our findings on these and other published genomes is in preparation. If you intend to use these sequences prior to our publication, please communicate with Drs. Muyembe-Tamfum, Ahuka-Mundeke, and Mbala-Kingebeni for coordination.
Data availability
References
Grubaugh ND, Gangavarapu K, Quick J, et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol 2019;20:8. 10.1186/s13059-018-1618-7
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 2013;30:772-80. 10.1093/molbev/mst010
Mbala-Kingebeni P, Pratt CB, Wiley MR, et al. 2018 Ebola virus disease outbreak in Equateur Province, Democratic Republic of the Congo: a retrospective genomic characterisation. Lancet Infect Dis 2019;19:641-7. 10.1016/S1473-3099(19)30124-0
Mbala-Kingebeni P, Aziza A, Di Paola N, et al. Medical countermeasures during the 2018 Ebola virus disease outbreak in the North Kivu and Ituri Provinces of the Democratic Republic of the Congo: a rapid genomic assessment. Lancet Infect Dis 2019;19:648-57. 10.1016/S1473-3099(19)30118-5
Quick, J., Grubaugh, N., Pullan, S. et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc 2017;12:1261–1276. https://doi.org/10.1038/nprot.2017.066
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014;30:1312-3. 10.1093/bioinformatics/btu033