Detection of the recurrent substitution Q677H in the spike protein of SARS-CoV-2 in cases descended from the lineage B.1.429
Christopher H. Tomkins-Tinch*‡1,2, Jillian Silbert*3, Katherine C. DeRuff*1, Katherine J. Siddle1,2, Adrianne Gladden-Young1, Amy Bronson4, John Marshall5, Zeynep Ozsoy Bean6, Kari Sholtes7,8, Kyle McQuade6, Anton Kary6, Kelly Krohn Bevill7, Clay King9, Michael Hughes5, Emma Leenerman5, Eric Parrie10, Emily Baron10, Megan E. Vodzak1, Gabrielle Gionet1, Gordon Adams1, Leah Pearlman1, Kian Sani1, Ivan Specht1, Andres Colubri1,11, Brittany A. Petros1,12,13, Christine Loreth1, Jacob E. Lemieux1,14, Stephen F. Schaffner1, Jeremy Luban1,15,16,17, Mark Schifferli18, Paul Cronan18, Olivia Glennon18, Kyle Oba18, Ben Fry18, Megan Doss19, Daniel J. Park1, Bronwyn L. MacInnis1, Pardis C. Sabeti‡1,2,17,20,21
1Broad Institute of MIT and Harvard, Cambridge, MA. 2Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, MA. 3Department of Systems Biology, Harvard Medical School, Boston, Massachusetts. 4Physician Assistant Program, Department of Kinesiology, Colorado Mesa University, Grand Junction, CO. 5Colorado Mesa University, Grand Junction, CO. 6Department of Biological Sciences, Colorado Mesa University, Grand Junction, CO. 7Department of Computer Science and Engineering, Colorado Mesa University, Grand Junction, CO. 8Department of Civil, Environmental, and Architectural Engineering, University of Colorado, Boulder, CO. 9Department of Mathematics and Statistics, Colorado Mesa University, Grand Junction, CO. 10COVIDCheck Colorado, LLC., Denver, CO. 11University of Massachusetts Medical School, Worcester, MA. 12Harvard-MIT Program in Health Sciences and Technology, Cambridge, MA. 13Harvard/MIT MD-PhD Program, Boston, MA. 14Division of Infectious Diseases, Massachusetts General Hospital, Boston, MA. 15Program in Molecular Medicine, University of Massachusetts Medical School, Worcester, MA. 16Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School, Worcester, MA. 17Massachusetts Consortium on Pathogen Readiness, Boston, MA. 18Fathom Information Design, Boston, MA. 19Warrior Diagnostics, Inc., Loveland, CO. 20Harvard T.H. Chan School of Public Health, Boston, MA. 21Howard Hughes Medical Institute, Chevy Chase, MD.
*These authors contributed equally to this work.
‡Corresponding authors
Abstract
Through a genomic surveillance program at Colorado Mesa University (CMU), we have identified a novel SARS-CoV-2 cluster descended from the variant of concern (VOC) lineage B.1.429 (CDC 2021) with an additional spike protein substitution at amino acid position 677, S:Q677H. The cluster of cases carries the core substitutions of the B.1.429 lineage (a.k.a. Cal.20C), which was first found in Southern California in September 2020 and has risen in frequency to comprise the majority of sequenced cases in California (“Outbreak.info”). They also contain the S:Q677H substitution, which has previously been observed in seven distinct lineages, raising interest in potential convergent evolution. Independently, both B.1.429 and lineages with S:Q677 substitutions have increased in prevalence(Hodcroft et al. 2021; Deng et al. 2021); observing S:Q677H on the B.1.429 background, therefore, warrants further surveillance of this newly emerged haplotype and highlights an urgent need to investigate the function of Q677 substitutions. Following the avian nomenclature developed in Hodcroft et al. for lineages bearing substitutions at spike position 677, we are referring to the B.1.429+S:Q677H lineage as “Peacock.”
Introduction
In partnership with Colorado Mesa University (CMU), a mid-sized university of 10,000 students in the US Mountain West, we have set up a genomic surveillance program to sequence SARS-CoV-2 from clinical excess specimens collected as part of the institutional surveillance testing program. The specimens sequenced were saliva samples from individual positive cases within the university community as well as collections of on-campus residential wastewater. Since November 2020, 20.2% of all positive test cases from students and faculty have been sequenced, for a total of 229 samples. Of these, 178 (78%) generated full genomes, representing 13.5% of published sequences on GenBank and GISAID from samples collected in Colorado.
From early summer 2020, CMU implemented contact tracing protocols and robust surveillance testing of individuals and wastewater. As of fall 2020, the university incorporated viral genomic sequencing of individual positive test cases and wastewater. The sampling strategy includes a tiered surveillance testing model with 56% of tests over the three months directed to close contacts of individuals who tested positive, symptomatic individuals, or individuals in residence halls with positive wastewater detected. Additionally, 50% of surveillance tests were dedicated to primary contacts of CMU members in the surrounding community. Wastewater effluent from residence halls was sampled twice weekly and tested via qPCR for the presence of SARS-CoV-2. Wastewater test results were used to guide inclusion in surveillance testing.
As part of our surveillance efforts, we have been investigating the emergence of known and novel variants of interest, such as the B.1.429 lineage. The B.1.429 lineage was first reported in January 2021 in California but is believed to have emerged in May 2020. It has increased in frequency in several states and has become the majority lineage in California and Arizona. The B.1.429 lineage descends from the broader 20C clade and is defined by four non-synonymous substitutions: three in the spike glycoprotein (S13I, W152C, L452R) and one in ORF1b (D1183Y). Functional investigation of B.1.429 found virus with this haplotype increased viral shedding in vivo, reduced neutralization by both convalescent and vaccine-induced antibodies, and increased infectivity and transmissibility (Deng et al. 2021). It has been reported that the investigational drug bamlanivimab, which has received emergency FDA approval for use in COVID-19 patients, has reduced neutralization of SARS-CoV-2 lineages bearing S:L452R (Starr et al. 2021). The B.1.429 lineage has now risen to account for the majority of all sequenced cases in California and Nevada (“Outbreak.info”). As of mid-March, it has been identified in SARS-CoV-2 sequences in every state except South Dakota.
An unrelated clade bearing S:Q677H was first detected in August 2020, and since then the S:Q677H substitution has independently developed in at least seven lineages across three clades (20A, 20B, 20G), with an additional lineage gaining S:Q677P (Hodcroft et al. 2021). These lineages were provisionally named Robin 1, Robin 2, Pelican, Yellowhammer, Bluebird, Quail, and Mockingbird. The independent acquisition of this mutation indicates it is a product of convergent evolution. Coupled with its rise to prominence, independent acquisition of S:Q677H suggests positive selection for S:Q677H. The S:Q677H mutation may affect cell entry due to its proximity to the polybasic cleavage site (Hodcroft et al. 2021). Its implications for human disease are currently unknown.
Methods
Total RNA was extracted from saliva samples collected from members of the CMU community. Presence of SARS-CoV-2 virus was confirmed using an RT-qPCR assay detecting the N1 gene of the virus. For wastewater specimens, CMU performed nucleic acid extraction immediately following collection. All positive and wastewater samples were sequenced using protocols described previously for preparing libraries with tiled amplicons using ARTIC v3 primers (Lagerborg et al. 2021; Tyson et al. 2020).
Viral genomes were assembled using the purpose-built viral-ngs v2.1.19.0 assembly pipelines (GitHub - broadinstitute/viral-pipelines: viral-ngs: complete pipelines) executed on the Terra platform (Terra). All workflows used are publicly available via the Dockstore Tool Registry Service (dockstore.org/organizations/BroadInstitute/collections/pgs). Phylogenetic trees were created via the sarscov2_nextstrain workflow using GISAID data for global context, and visualized using the Nextstrain tool Auspice (Elbe and Buckland-Merrett 2017; Hadfield et al. 2018). Single-nucleotide variants were visualized using snipit (O’Toole).
Results
Of 229 samples we collected from October through February, 93 were found to contain the S:Q677H mutation. These 93 samples belong to the following lineages: Robin 1 (n=22), Robin 2 (n=2), Pelican (n=21), and Quail (n=1). Of the 47 samples not perfectly concordant with the haplotypes of lineages as defined in Hodcroft et al., 15 had gaps in sequencing coverage but otherwise are consistent with the Robin 1, Robin 2, and Quail lineages. However, 32 samples clearly belonged to a different lineage, whose characteristic mutations are defined in Table 1. All 32 are members of clade 20C and are descendants of the B.1.429 lineage; we are referring to this lineage as “Peacock.”
Table 1: Amino acid changes characteristic of B.1.429 v. Peacock
Shown in bold are the derived substitutions in Peacock, beyond those which define B.1.429. Amino acid changes in italic are B.1.429-derived substitutions observed in published data for samples collected in California.
| Region | Amino acid changes characteristic of B.1.429 | Amino acid changes characteristic of Peacock (N=32) |
|---|---|---|
| ORF1a | T265I,I4205V | T265I, F2827L,I4205V,V3367I |
| ORF1b | P314L, D1183Y | P314L, D1183Y |
| S | S13I, W152C, L452R, D614G | S13I, W152C*, L452R*, D614G*, Q677H |
| ORF3a | Q57H | A23V, Q57H |
| N | T205I | P142S, T205I*, M234I* |
| ORF8 | V100L | |
| * Due to gaps in sequencing coverage, some substitutions could not be called for a minority of samples but are presumed to be present based on complementary substitutions in the shared lineage-defining haplotype. |
Table 2: Amino acid changes characteristic of Peacock and their corresponding nucleotide substitutions, relative to NC_045512.2 (Wuhan-Hu-1)
| Amino acid change | Nucleotide substitution |
|---|---|
| ORF1a:F2827L | T8744C |
| ORF1a:V3367I | G10364A |
| S:Q677H | G23593C |
| ORF3a:A23V | C25460T |
| N:P142S | C28697T |
| N:M234I | G28975T |
| ORF8:V100L | G28191T |
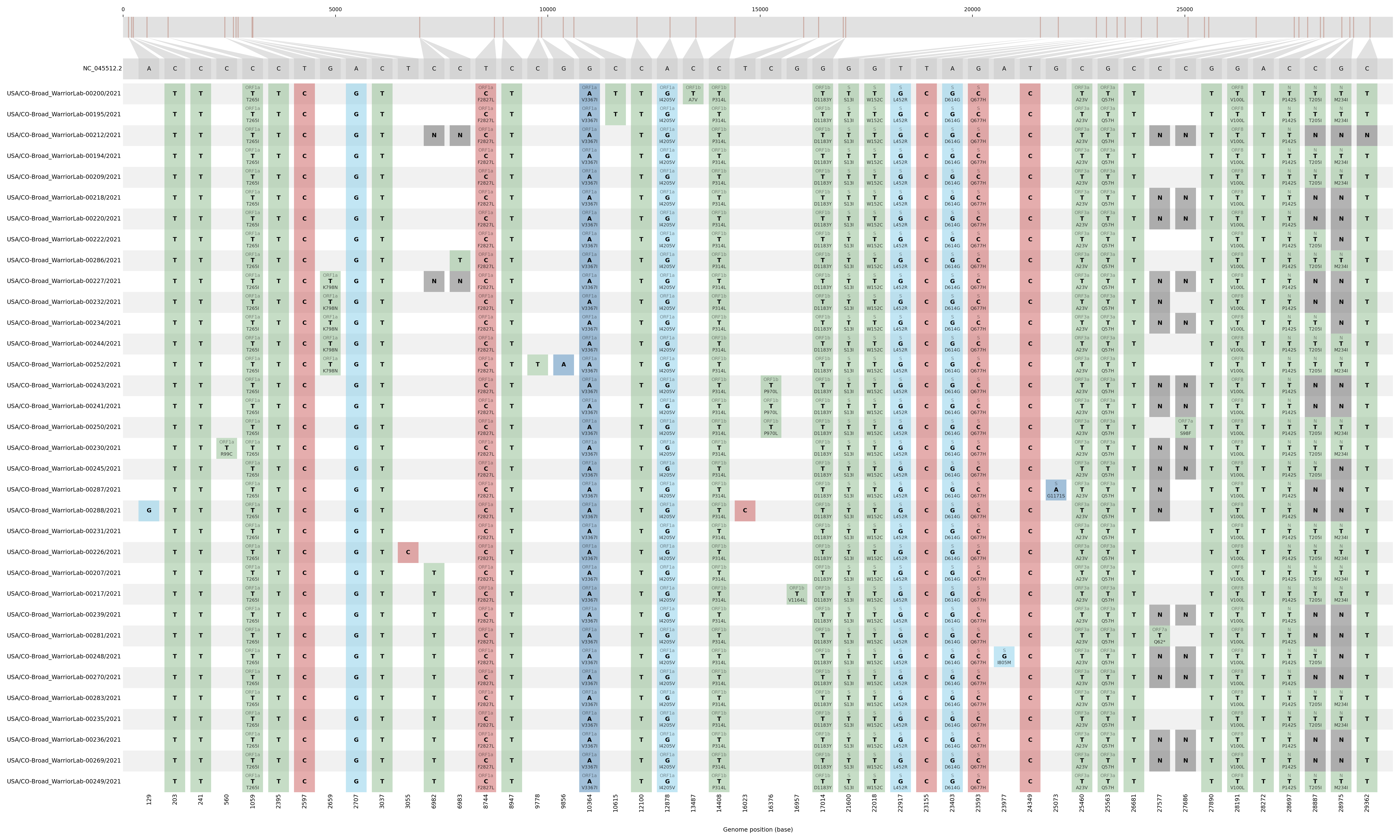
Peacock genome counts on campus over time
The Peacock cluster was most prevalent in early February, but Peacock cases were still detected in mid- to late February. At its apparent peak, Peacock samples represented 80% of sequenced positive cases in a given day. Total positive SARS-CoV-2 cases on campus have decreased since January, as a result, data for February are sparse.

Single-nucleotide polymorphisms in this cluster, relative to NC_045512.2 (Wuhan-Hu-1)
Phylogenetic tree of Peacock cluster in context: very recent, derived from a handful of B.1.429 cases samples in California
A: The cluster of Peacock cases, shown in yellow, observed in Colorado at the tip of the 20C clade in the context of contextual sequence data weighted toward sequences from the US Mountain West, followed by sequences from the United States, and then international sources. B: The Peacock cases in the context of the broader B.1.429 lineage. C: The clade of B.1.429+S:Q677H by date of collection. There are four distinct clusters, three descended from the initial cluster. Each cluster has cases close in time and genetic distance, exhibiting star phylogenies consistent with clonal amplification events. The time to most recent common ancestor (tMRCA) of the parent cluster was inferred to be 2021-01-19 (95% CI: 2021-01-17 to 2021-01-25). The earliest sequenced samples in the B.1.429+S:Q677H clade were collected on January 27, approximately one week after the inferred tMRCA. This data can be explored online: https://auspice.broadinstitute.org/sars-cov-2/us-mountain-west-grouping-weighted/20210309

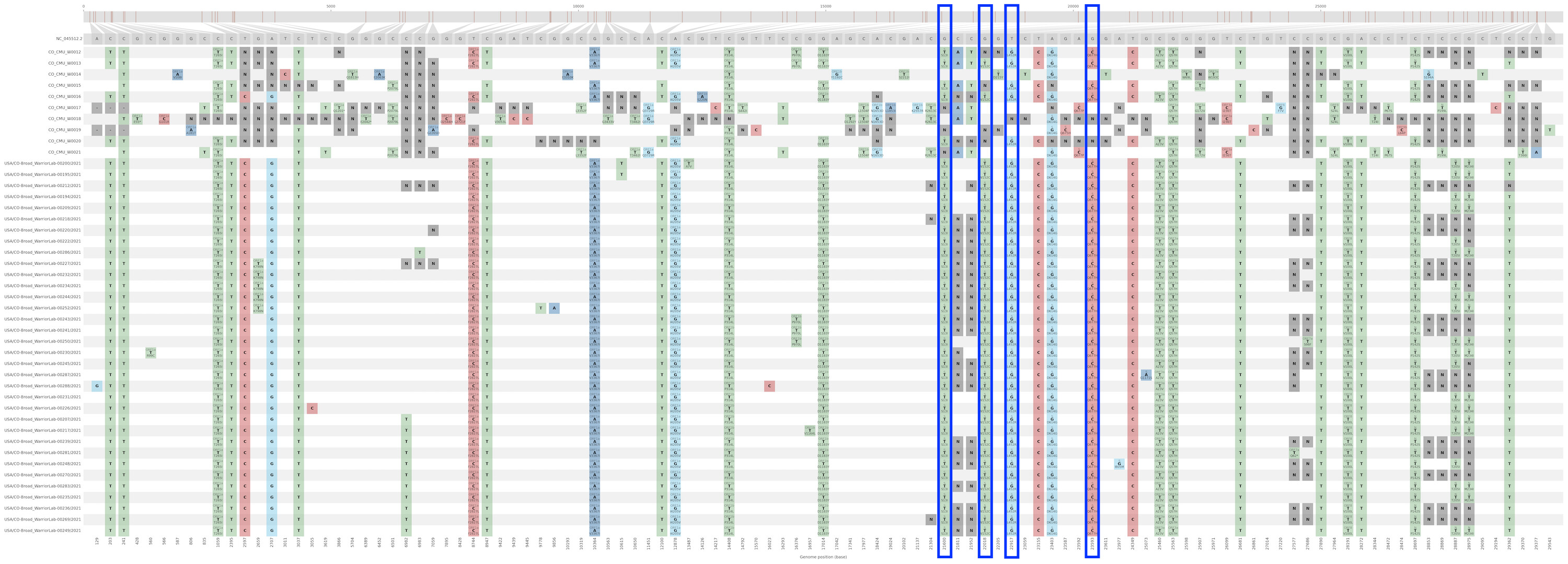
Single-nucleotide polymorphisms present in this cluster, including wastewater samples, relative to NC_045512.2
Based on limited data from ten wastewater samples (top ten rows) collected from nine residence halls across three days, the B.1.429+S:Q677H cluster was confidently identified as the majority haplotype in the wastewater from three residence halls.Outlined in blue are the spike substitutions S:S13I, S:L452R, S:W152C, and S:Q677H. W0012 and W0013 were collected from the same building.
Discussion
The B.1.429 lineage was first detected in Colorado on 2020-12-15 (MW550638.1; EPI_ISL_903861). To our knowledge based on data available on GISAID as of 2021-03-19, the cluster we have identified includes the first sequences in Colorado with the B.1.429+S:Q677H haplotype. In less than a month, the Peacock lineage emerged and became prominent among positive SARS-CoV-2 cases on the CMU campus. The sudden and dramatic increase in prevalence of Peacock-lineage cases may be an artifact of clonal amplification (“superspreading”) events on the CMU campus, consistent with the polytomies observed in the phylogeny of the cluster. However, it should be noted that outside the context of this cluster both the B.1.429 lineage and the S:Q677H mutation rapidly rose in prevalence, implying that either may confer relevant selective advantages for the virus (Deng et al. 2021; Hodcroft et al. 2021). The convergent evolution of S:Q677H is consistent with positive selection for the S:Q677H mutation.
This cluster was quickly recognized as notable. It is not the first occurrence of this constellation of substitutions; the first published B.1.429-lineage sequence bearing S:Q677H was from a sample collected in Southern California on 2020-12-18 (GenBank: MW485144.1, GISAID: EPI_ISL_832113). Clusters from these sequences are not present in the published data, though unsampled case clusters may have occurred. Several additional cases were sequenced from California and Arizona before the detection of the cluster in Colorado described here (Peng et al. 2021; Elbe and Buckland-Merrett 2017). The first two Peacock cases from the Colorado cluster described here were collected on 2021-01-27, approximately one week after the inferred tMRCA for the cluster, 2021-01-19. We believe that the earliest two cases of the cluster descend from an unsampled case not included in our dataset. The cluster was first identified by sequencing within two weeks of the first case: in this time frame the sample was tested by qPCR, identified as positive, shipped to our sequencing facility, sequenced, analyzed, and flagged as different and notable.
It has been shown that quantitation of SARS-CoV-2 in wastewater can be a leading indicator of case counts (Wu et al. 2020). We are unable to use the wastewater sequenced here to provide evidence for earlier presence of Peacock on campus as the earliest samples from this CMU cohort are from February 12, after the start of the cluster. Moving forward, we will examine whether wastewater sequencing can detect variants of interest or concern in advance of detection by sequencing of positive cases.
The Peacock lineage is the first VOC to include S:Q677H, though the Variant of Interest (VOI) lineage B.1.525 also includes S:Q677H (CDC 2021). While the functional implications of S:Q677H on the B.1.429 haplotype are presently unknown, the repeated appearance of this mutation in multiple lineages suggests that Peacock is a lineage that warrants monitoring.
Data Availability
An interactive phylogeny including this cluster is available at the following URL:
https://auspice.broadinstitute.org/sars-cov-2/us-mountain-west-grouping-weighted/20210309
Sequences produced from samples collected at CMU are associated with NCBI BioProject PRJNA622837; contextual sequences used in the Phylogenetic tree are from GISAID as of 2021-03-09. Sequences from wastewater samples had insufficient coverage to meet quality standards for release.
The Peacock sequences are available on GenBank under the following accessions:
| Sequence ID | GenBank Accession | GISAID ID |
|---|---|---|
| USA-CO-Broad_WarriorLab-00194-2021 | MW617686 | EPI_ISL_1011654 |
| USA-CO-Broad_WarriorLab-00195-2021 | MW617687 | EPI_ISL_1011655 |
| USA-CO-Broad_WarriorLab-00200-2021 | MW617692 | EPI_ISL_1011660 |
| USA-CO-Broad_WarriorLab-00207-2021 | MW617699 | EPI_ISL_1011667 |
| USA-CO-Broad_WarriorLab-00209-2021 | MW617701 | EPI_ISL_1011669 |
| USA-CO-Broad_WarriorLab-00212-2021 | MW617704 | EPI_ISL_1011672 |
| USA-CO-Broad_WarriorLab-00217-2021 | MW617708 | EPI_ISL_1011676 |
| USA-CO-Broad_WarriorLab-00218-2021 | MW617709 | EPI_ISL_1011677 |
| USA-CO-Broad_WarriorLab-00220-2021 | MW749875 | EPI_ISL_1253886 |
| USA-CO-Broad_WarriorLab-00222-2021 | MW749877 | EPI_ISL_1253888 |
| USA-CO-Broad_WarriorLab-00226-2021 | MW749879 | EPI_ISL_1253890 |
| USA-CO-Broad_WarriorLab-00227-2021 | MW749880 | EPI_ISL_1253891 |
| USA-CO-Broad_WarriorLab-00230-2021 | MW749883 | EPI_ISL_1253894 |
| USA-CO-Broad_WarriorLab-00231-2021 | MW749884 | EPI_ISL_1253895 |
| USA-CO-Broad_WarriorLab-00232-2021 | MW749885 | EPI_ISL_1253896 |
| USA-CO-Broad_WarriorLab-00234-2021 | MW749887 | EPI_ISL_1253898 |
| USA-CO-Broad_WarriorLab-00235-2021 | MW749888 | EPI_ISL_1253899 |
| USA-CO-Broad_WarriorLab-00236-2021 | MW749889 | EPI_ISL_1253900 |
| USA-CO-Broad_WarriorLab-00239-2021 | MW749891 | EPI_ISL_1253902 |
| USA-CO-Broad_WarriorLab-00241-2021 | MW749892 | EPI_ISL_1253903 |
| USA-CO-Broad_WarriorLab-00243-2021 | MW749894 | EPI_ISL_1253905 |
| USA-CO-Broad_WarriorLab-00244-2021 | MW749895 | EPI_ISL_1253906 |
| USA-CO-Broad_WarriorLab-00245-2021 | MW749896 | EPI_ISL_1253907 |
| USA-CO-Broad_WarriorLab-00248-2021 | MW749898 | EPI_ISL_1253909 |
| USA-CO-Broad_WarriorLab-00249-2021 | MW749899 | EPI_ISL_1253910 |
| USA-CO-Broad_WarriorLab-00250-2021 | MW749900 | EPI_ISL_1253911 |
| USA-CO-Broad_WarriorLab-00252-2021 | MW749902 | EPI_ISL_1253913 |
| USA-CO-Broad_WarriorLab-00269-2021 | MW749916 | EPI_ISL_1253927 |
| USA-CO-Broad_WarriorLab-00272-2021 | MW749918 | EPI_ISL_1253929 |
| USA-CO-Broad_WarriorLab-00283-2021 | MW749924 | EPI_ISL_1253935 |
| USA-CO-Broad_WarriorLab-00286-2021 | MW749927 | EPI_ISL_1253938 |
| USA-CO-Broad_WarriorLab-00287-2021 | MW749928 | EPI_ISL_1253939 |
| USA-CO-Broad_WarriorLab-00288-2021 | MW749929 | EPI_ISL_1253940 |
Acknowledgements
The authors wish to acknowledge the students and staff of Colorado Mesa University for participating in this work and for their commitment to the health of their community, and thank Lydia A. Krasilnikova, Bennett M. Shaw, and others in the Sabeti Lab for their assistance, editing, feedback, and collaboration. We gratefully acknowledge the laboratories and researchers who made SARS-CoV-2 genomes available on GISAID, which we utilized in our phylogenetic analyses. This work was sponsored in part by the Howard Hughes Medical Institute Investigator award to P.C.S., the National Institute of Allergy and Infectious Diseases (U19AI110818 to P.C.S), the Centers for Disease Control Broad Agency Announcement (75D30120C09605 to BLM), National Science Foundation Graduate Research Fellowships (Grant No. 1745303) to C.T.-T. and J.S., a Hertz fellowship to J.S, and award Number T32GM007753 from the National Institute of General Medical Sciences to B.A.P. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Conflicts of interest
Pardis C. Sabeti is a co-founder and shareholder of Sherlock Biosciences and is a non-executive board member and shareholder of Danaher Corporation.
Ethical Statement
The study was conducted at the Broad Institute with approval from the MIT Institutional Review Board under Protocol #1612793224.
References
CDC. 2021. “SARS-CoV-2 Variants.” March 16, 2021. https://www.cdc.gov/coronavirus/2019-ncov/cases-updates/variant-surveillance/variant-info.html.
Deng, Xianding, Miguel A. Garcia-Knight, Mir M. Khalid, Venice Servellita, Candace Wang, Mary Kate Morris, Alicia Sotomayor-Gonzalez, et al. 2021. “Transmission, Infectivity, and Antibody Neutralization of an Emerging SARS-CoV-2 Variant in California Carrying a L452R Spike Protein Mutation.” bioRxiv. medRxiv. https://doi.org/10.1101/2021.03.07.21252647.
Elbe, Stefan, and Gemma Buckland-Merrett. 2017. “Data, Disease and Diplomacy: GISAID’s Innovative Contribution to Global Health.” Global Challenges (Hoboken, NJ) 1 (1): 33–46.
Hadfield, James, Colin Megill, Sidney M. Bell, John Huddleston, Barney Potter, Charlton Callender, Pavel Sagulenko, Trevor Bedford, and Richard A. Neher. 2018. “Nextstrain: Real-Time Tracking of Pathogen Evolution.” Bioinformatics 34 (23): 4121–23.
Hodcroft, Emma B., Daryl B. Domman, Daniel J. Snyder, Kasopefoluwa Oguntuyo, Maarten Van Diest, Kenneth H. Densmore, Kurt C. Schwalm, et al. 2021. “Emergence in Late 2020 of Multiple Lineages of SARS-CoV-2 Spike Protein Variants Affecting Amino Acid Position 677.” medRxiv : The Preprint Server for Health Sciences, February. https://doi.org/10.1101/2021.02.12.21251658.
Lagerborg, Kim A., Erica Normandin, Matthew R. Bauer, Gordon Adams, Katherine Figueroa, Christine Loreth, Adrianne Gladden-Young, et al. 2021. “DNA Spike-Ins Enable Confident Interpretation of SARS-CoV-2 Genomic Data from Amplicon-Based Sequencing.” Cold Spring Harbor Laboratory. https://doi.org/10.1101/2021.03.16.435654.
O’Toole, Áine. n.d. Snipit. Github. Accessed March 18, 2021. https://github.com/aineniamh/snipit.
“Outbreak.info.” Accessed March 18, 2021a. https://outbreak.info/situation-reports?pango=B.1.429; Accessed March 18, 2021b. https://outbreak.info/situation-reports?pango=B.1.429&loc=USA_US-NV&loc=USA_US-CA&selected=USA_US-NV.
Peng, James, Sabrina A. Mann, Anthea M. Mitchell, Jamin Liu, Matthew T. Laurie, Sara Sunshine, Genay Pilarowski, et al. 2021. “Estimation of Secondary Household Attack Rates for Emergent SARS-CoV-2 Variants Detected by Genomic Surveillance at a Community-Based Testing Site in San Francisco.” medRxiv : The Preprint Server for Health Sciences, March. https://doi.org/10.1101/2021.03.01.21252705.
Starr, Tyler N., Allison J. Greaney, Adam S. Dingens, and Jesse D. Bloom. 2021. “Complete Map of SARS-CoV-2 RBD Mutations That Escape the Monoclonal Antibody LY-CoV555 and Its Cocktail with LY-CoV016.” bioRxiv : The Preprint Server for Biology, February. https://doi.org/10.1101/2021.02.17.431683.
Tyson, John R., Phillip James, David Stoddart, Natalie Sparks, Arthur Wickenhagen, Grant Hall, Ji Hyun Choi, et al. 2020. “Improvements to the ARTIC Multiplex PCR Method for SARS-CoV-2 Genome Sequencing Using Nanopore.” bioRxiv : The Preprint Server for Biology, September. https://doi.org/10.1101/2020.09.04.283077.
Wu, Fuqing, Amy Xiao, Jianbo Zhang, Katya Moniz, Noriko Endo, Federica Armas, Richard Bonneau, et al. 2020. “SARS-CoV-2 Titers in Wastewater Foreshadow Dynamics and Clinical Presentation of New COVID-19 Cases.” medRxiv : The Preprint Server for Health Sciences, June. https://doi.org/10.1101/2020.06.15.20117747.
gisaid_author_acknowledgement.tsv.zip (26.0 KB)