Detection of Spike Mutations; D80G, T95I, G142D, △144, N439K, E484K, P681H, I1130V, and D1139H, in B.1.1.482 Lineage (AV.1) Samples from South Yorkshire, UK.
+Sharon Hsu1,2 and +Matthew D Parker2,3,4, Benjamin B Lindsey1,5, Amy State5, Peijun Zhang1, Benjamin H Foulkes1, Stavroula F Louka1, Paige Wolverson1, Marta Gallis1, Hailey R Hornsby1, Max Whiteley1, Samantha E Hansford1, Nasar Ali5, Oscar Shepherdson5, Salma Afsar5, Katie Johnson5, Alexander J Keeley1,5, Joe Heffer6, Nikki Smith1, David G Partridge5, Alison Cope5, Michael Ankcorn5, Mohammad Raza5, Cariad Evans5, The COVID-19 UK Genomics Consortium7, Thushan I de Silva1,5*
1 The Florey Institute for Host-Pathogen Interactions & Department of Infection, Immunity and Cardiovascular Disease, Medical School, University of Sheffield, Sheffield, UK.
2 Sheffield Bioinformatics Core, The University of Sheffield
3 Sheffield Biomedical Research Centre, The University of Sheffield
4 The Department of Neuroscience/Neuroscience Institute, The University of Sheffield
5 Sheffield Teaching Hospitals NHS Foundation Trust, Sheffield, UK.
6 IT Services, The University of Sheffield, UK.
7 https://www.cogconsortium.uk, a full list of consortium names and affiliations is located here
+ These authors contributed equally to this work
* Correspondence: [email protected]
Summary
Here we report the discovery of 24 recent B.1.1.482 pango lineage SARS-CoV-2 viruses in South Yorkshire that have acquired approximately 23 additional mutations when compared to the majority of other samples of this lineage. Nine of these additional mutations are found in the spike gene and six represent changes which are found in variants of interest and concern across the world and/or associated with reduced antibody binding (T95I, G142D, △144, N439K, E484K, P681H). Similar mutation profiles were also seen in B.1.1.482 samples from other regions in the UK. We recommended the monitoring of samples of this lineage which is now designated AV.1.
Background
De-centralised rapid sequencing and analysis of SARS-CoV-2 genomes has underpinned our ability to monitor the emergence and spread of SARS-CoV-2 variants of concern in the UK. As part of COG-UK (COVID-19 Genomics UK (COG-UK) 2020) the Sheffield COVID-19 Genomics Group are sequencing SARS-CoV-2 positive samples from hospitals throughout Yorkshire and The Humber (pillar 1 testing) to ensure local NHS trusts and public health teams are delivered timely information on the lineage composition of SARS-CoV-2 circulating in the community.
Variants of concern (VOC) can be more transmissible (Davies et al. 2020), result in greater disease severity (Davies et al. 2021), and have reduced sensitivity to antibody neutralisation (Wang et al. 2021). These so called VOC often harbour multiple mutations in the spike protein which may result in reduced effectiveness of SARS-CoV-2 vaccines. We must therefore track current and identify new variants rapidly to ensure an appropriate public health response. B.1.1.7 pango lineage SARS-CoV-2 has dominated the epidemic in South Yorkshire since early January 2021 (Parker et al. 2021) and non-B.1.1.7 lineage SARS-CoV-2 viruses are rare. Here we describe the discovery of B.1.1.482 viruses in South Yorkshire, UK, with a constellation of spike gene mutations not found before in this lineage.
Results & Discussion
Samples of B.1.1.482 Lineage from South Yorkshire With Additional Shared Mutations
Routine surveillance sequencing was performed on four nose and throat swab samples (SHEF-10DA497, SHEF-10E58AC, SHEF-10DC3F2, SHEF-10D9A3C) that tested SARS-CoV-2 positive on RT-PCR at Sheffield Teaching Hospitals NHS Foundation Trust. SHEF-10DA497 was collected on the 28th of April 2021, both SHEF-10E58AC and SHEF-10DC3F2 were collected on the 30th April 2021 and SHEF-10D9A3C was collected on 7th May 2021. Sequencing was performed using the ARTIC network protocol (V3-LoCost)(“Artic Network” 2020) and an Oxford Nanopore GridION X5. A consensus genome was produced using the ARTIC bioinformatics pipeline (v1.1.1). All four sequences were classified into the B.1.1.482 pango lineage (Rambaut et al. 2020) by pangolin (version 2.3.8, pangoLEARN 2021-04-28).
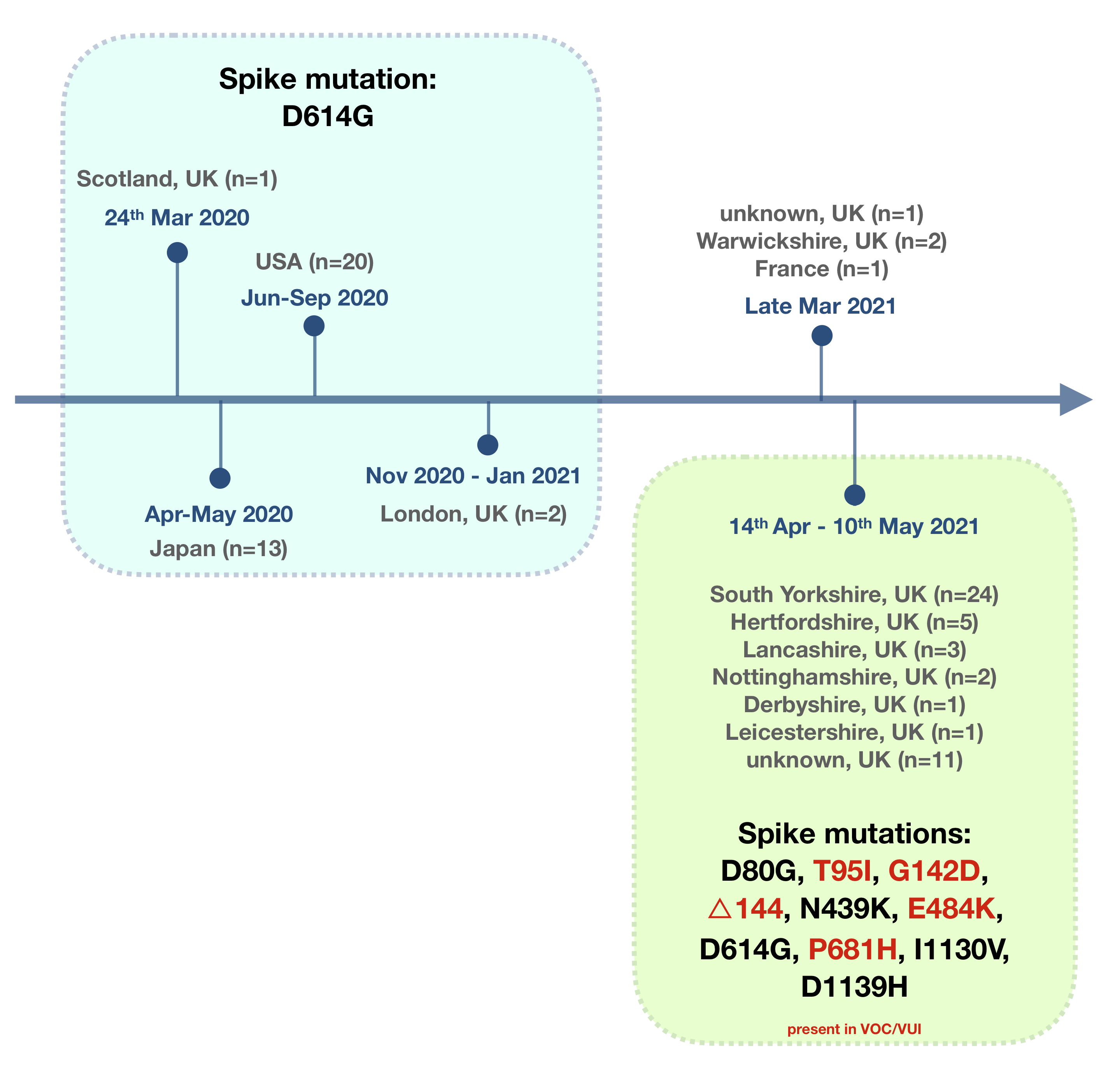
B.1.1.482 was 1st seen on 24th March 2020 (cov-lineages.org) in Scotland and has been found in the USA, Japan, and more recently France and now England. Further investigation of SHEF-10DA497 revealed a total of 31 amino acid changes with respect to MN908947.3 (Table 1), with ten of these located within the spike gene. This constellation of mutations is also found in the other three Sheffield samples (SHEF-10E58AC, SHEF-10DC3F2, SHEF-10D9A3C), 20 South Yorkshire pillar 2 (community) samples, and samples from 5 other regions in England (A full list of samples used in our analysis can be found here). Pillar 2 samples were sequenced by the Wellcome Trust Sanger Institute and other COG-UK sequencing centres as part of the UKs community sequencing programme. Older B.1.1.482 samples from the USA and Japan share only a small subset of these mutations (Table 1 in bold).
| Genomic Region | Protein | Amino Acid |
|---|---|---|
| ORF1a | NSP1 | D75G |
| ORF1a | NSP2 | G339S, A411V |
| ORF1a | NSP3 | H342Y, P822L, K927I, N1587S |
| ORF1a | NSP4 | A446V |
| ORF1a | NSP6 | △106-108 |
| ORF1b | NSP12 | P323L, S434A |
| ORF1b | NSP13 | T481M |
| ORF1b | NSP14 | A119V |
| S | Spike | D80G, T95I, G142D, △144, N439K, E484K, D614G, P681H, I1130V, D1139H |
| M | Membrane | A63T, H125Y |
| N | Nucleocapsid | I157V, R203K, G204R |
Table 1. Mutations common (n=28) to B.1.1.482 samples from the South Yorkshire Region.
Mutations overlapping B.1.1.482 from North America and Asia indicated in bold.
A maximum likelihood phylogenetic tree (Figure 1) was constructed to reveal the heterogeneity within the current B.1.1.482 lineage. A high degree of divergence was observed between the B.1.1.482 samples from the USA and Japan, and those from France and South Yorkshire, UK. Importantly, three recent B.1.1.28 samples (QEUH-148F257, QEUH-14C1E55, QEUH-144D7A1) appear to form an intermediate cluster between the South Yorkshire samples and the sample from France suggesting that this could be a common ancestor of the two clusters.
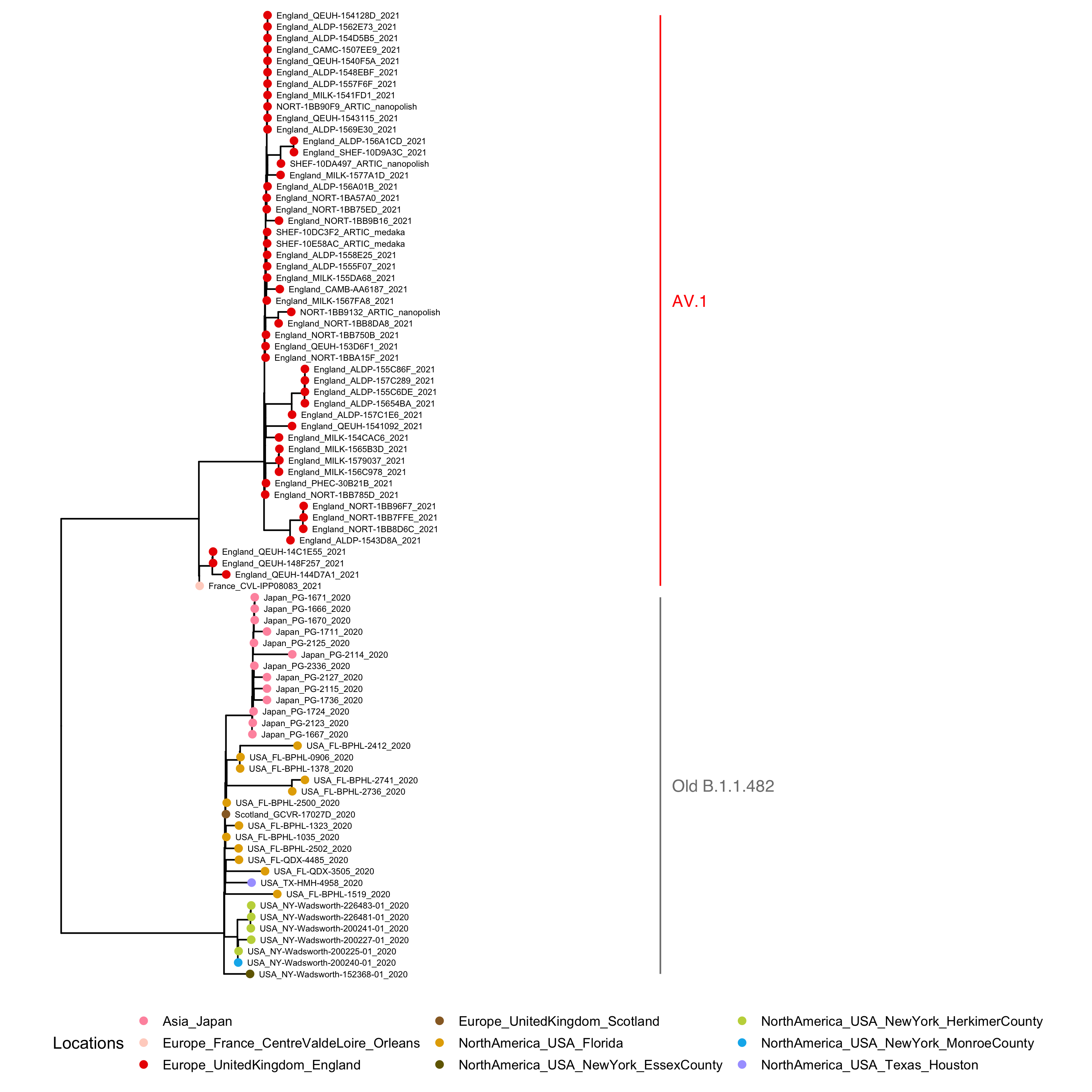
Figure 1. Maximum likelihood Phylogenetic Tree of B.1.1.482 and B.1.1.482-Related Samples.
A multiple sequence alignment was performed on 85 B.1.1.482 and B.1.1.482-Related Samples using MAFFT v.7.477. The maximum likelihood phylogenetic tree was produced using IQ-TREE v.2.1.2 with GTR + G model and 1,000 ultrafast bootstrap replicates. Red tips denote samples from England. B.1.1.482 samples with additional mutations are now designated as AV.1 with the latest pangoLEARN (2021-05-12).
The mutations contained within the B.1.1.482 and the three B.1.1.28 samples were visualised using snipit (O’Toole 2021). From this schematic it is clear that although the South Yorkshire B.1.1.482 samples share key B.1.1.482 lineage mutations, the SARS-CoV-2 viruses infecting these individuals have acquired a significant amount of further mutations which are shared between them. The French sample and the three B.1.1.28 (Figure 2 - bottom) samples all share a similar mutation profile to the South Yorkshire B.1.1.482 samples, with a few exceptions.
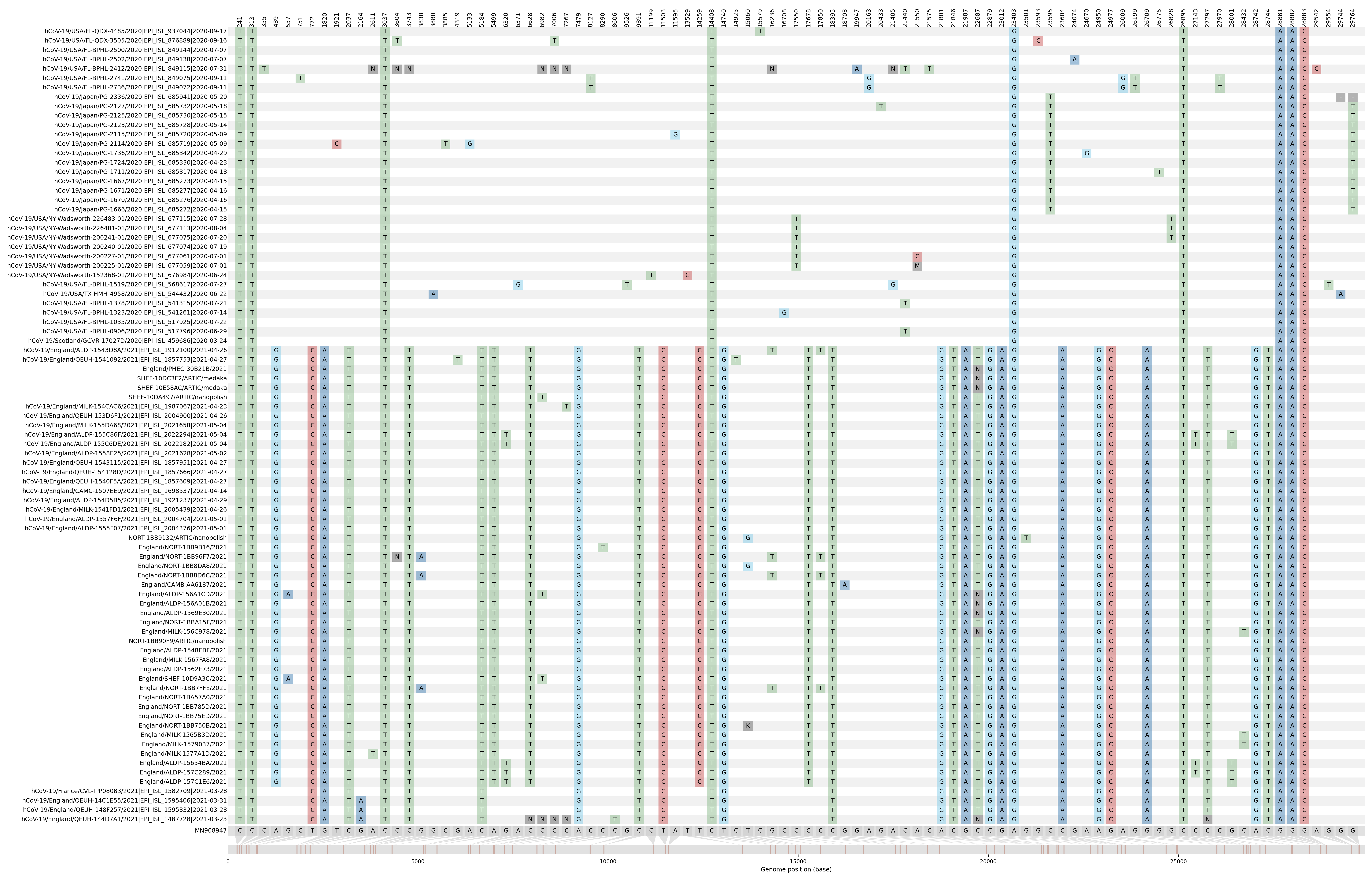
Figure 2. Mutation profile of the B.1.1.482 and related samples.
Comparison of mutations found in each of the B.1.1.482 samples visualised with snipit (O’Toole 2021) using a multiple sequence alignment generated with MAFFT (v.7.477). MN908947 was used as the reference sequence.
Spike Gene Mutations Also Found in Variants of Concern or Variants Under Investigation
Several of the mutations detected in the South Yorkshire B.1.1.482 samples in the Spike gene are also found in lineages that have been designated as Variants of Concern (VOC), or Variants Under Investigation (VUI) by Public Health England (https://github.com/phe-genomics/variant_definitions and https://www.gov.uk/government/publications/covid-19-variants-genomically-confirmed-case-numbers/variants-distribution-of-cases-data). In addition to the now ubiquitous D614G spike mutation (Korber et al. 2020), five other spike mutations are also found in lineages designated as a VOC or VUI:
T95I has been found in samples of the pango lineages B.1.1.318 (VUI-21FEB-04), B.1.526 (being monitored) and more recently in several B.1.617.1 (VUI-21APR-01) and B.1.617.2 (VOC-21APR-02) viruses. The impact of this mutation is currently unknown.
G142D is found in samples of the pango lineages B.1.617.1, B.1.617.2 and B.1.617.3. There is also a deletion at position 142 in P.3 (VUI-21MAR-02) as part of △141 - 142. G142D may reduce recognition of some monoclonal antibodies (Suryadevara et al. 2021; McCallum et al. 2021).
△144 is found in B.1.1.7, B.1.1.318, B.1.525 (VUI-21FEB-03) lineages and has been previously shown to reduce binding to monoclonal sera (McCallum et al. 2021).
E484K is located in the receptor binding ridge of the spike protein and is found in many lineages including B.1.351 (VOC-20DEC-02), P.1 (VOC-21JAN-02), A.23.1 (VUI-21FEB-01), B.1.525, B.1.1.318, P.2 (VUI-21JAN-01), B.1.324.1, a subclade of B.1.526, and P.3 (VUI-21MAR-02). This mutation reduces binding to polyclonal sera (Greaney, Loes, et al. 2021b) and escapes treatment with the antibody REGN10933 (Starr et al. 2021) which is part of the REGN-COV2 cocktail. It also results in escape from class 2 antibodies (Greaney, Starr, et al. 2021).
P681H is located adjacent to the spike protein furin cleavage site and is found in B.1.1.7 (VOC-20DEC-01), B.1.1.318 and P.3 (VUI-21MAR-02), and P681R is found in A23.1 and all B.1.617 lineages. P681H has been shown to enhance cleavage of spike (Brown et al. 2021). The impact of this increased efficiency in cleavage is not clear but is one hypothesis to explain the enhanced transmissibility of B.1.1.7.
Other Spike Mutations of Interest
N439K is not currently found in any VOC or VUI but has been shown to increase SARS-CoV-2 spike protein’s affinity to Ace2 (Starr et al. 2020). This mutation confers resistance to several monoclonal antibodies, with varying effect on escape from polyclonal sera (Greaney, Loes, et al. 2021a; Thomson et al. 2021).
D80G is seen in B.1.526.1 which is currently circulating in the USA, but not yet defined as a VUI or VOC by PHE. D80A is found in B.1.351. The impact of these mutations is unknown.
Other Mutations of Interest
In addition to mutations in the spike gene, △106-108 in the NSP6 open reading frame is a defining mutation of the B.1.1.7 lineage.
Conclusion
The recent B.1.1.482 samples from South Yorkshire described here have acquired a significant number of mutations when compared to other SARS-CoV-2 viruses of the same lineage. Similar samples were also found in other regions in England, UK. Three UK B.1.1.28 samples are believed to be related to this cluster. Independent sequencing of samples at The Wellcome Trust Sanger Institute (from multiple UK testing centres), The Institut Pasteur, and the COVID-19 Genomics Group in Sheffield and others rules out the possibility of within sequencing centre contamination or other unexplained centre specific phenomena. This sub-lineage of B.1.1.482 has now been designated AV.1 (https://github.com/cov-lineages/pango-designation/issues/73). Because of the number of spike gene mutations also found in VOC or VUI, we recommended that this lineage is monitored closely for possible increased community transmission and functional studies undertaken to understand the impact on antibody recognition.
References
“Artic Network.” 2020. November 5, 2020. https://artic.network/ncov-2019.
Brown, Jonathan C., Daniel H. Goldhill, Jie Zhou, Thomas P. Peacock, Rebecca Frise, Niluka Goonawardane, Laury Baillon, et al. 2021. “Increased Transmission of SARS-CoV-2 Lineage B.1.1.7 (VOC 2020212/01) Is Not Accounted for by a Replicative Advantage in Primary Airway Cells or Antibody Escape.” bioRxiv. https://doi.org/10.1101/2021.02.24.432576.
COVID-19 Genomics UK (COG-UK) [email protected]. 2020. “An Integrated National Scale SARS-CoV-2 Genomic Surveillance Network.” The Lancet. Microbe 1 (3): e99–100.
Davies, Nicholas G., Sam Abbott, Rosanna C. Barnard, Christopher I. Jarvis, Adam J. Kucharski, James Munday, Carl A. B. Pearson, et al. 2020. “Estimated Transmissibility and Severity of Novel SARS-CoV-2 Variant of Concern 202012/01 in England.” bioRxiv. medRxiv. https://doi.org/10.1101/2020.12.24.20248822.
Davies, Nicholas G., Christopher I. Jarvis, W. John Edmunds, Nicholas P. Jewell, Karla Diaz-Ordaz, and Ruth H. Keogh. 2021. “Increased Hazard of Death in Community-Tested Cases of SARS-CoV-2 Variant of Concern 202012/01.” medRxiv : The Preprint Server for Health Sciences, February. https://doi.org/10.1101/2021.02.01.21250959.
Greaney, Allison J., Andrea N. Loes, Katharine H. D. Crawford, Tyler N. Starr, Keara D. Malone, Helen Y. Chu, and Jesse D. Bloom. 2021a. “Comprehensive Mapping of Mutations to the SARS-CoV-2 Receptor-Binding Domain That Affect Recognition by Polyclonal Human Serum Antibodies.” bioRxiv. https://doi.org/10.1101/2020.12.31.425021.
Greaney, Allison J; Loes, Andrea N; Crawford, Katharine H D; Starr, Tyler N; Malone, Keara D; Chu, Helen Y; Bloom, Jesse D. 2021b. “Comprehensive Mapping of Mutations in the SARS-CoV-2 Receptor-Binding Domain That Affect Recognition by Polyclonal Human Plasma Antibodies.” Cell Host & Microbe 29 (3): 463–76.e6.
Greaney, Allison J., Tyler N. Starr, Christopher O. Barnes, Yiska Weisblum, Fabian Schmidt, Marina Caskey, Christian Gaebler, et al. 2021. “Mutational Escape from the Polyclonal Antibody Response to SARS-CoV-2 Infection Is Largely Shaped by a Single Class of Antibodies.” bioRxiv : The Preprint Server for Biology, March. https://doi.org/10.1101/2021.03.17.435863.
Korber, Bette, Will M. Fischer, Sandrasegaram Gnanakaran, Hyejin Yoon, James Theiler, Werner Abfalterer, Nick Hengartner, et al. 2020. “Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus.” Cell 182 (4): 812–27.e19.
McCallum, Matthew, Anna De Marco, Florian A. Lempp, M. Alejandra Tortorici, Dora Pinto, Alexandra C. Walls, Martina Beltramello, et al. 2021. “N-Terminal Domain Antigenic Mapping Reveals a Site of Vulnerability for SARS-CoV-2.” Cell 184 (9): 2332–47.e16.
O’Toole, Áine. Snipit. Github. Accessed May 6, 2021. GitHub - aineniamh/snipit: snipit: summarise snps relative to your reference sequence.
Parker, Matthew D., Benjamin B. Lindsey, Dhruv R. Shah, Sharon Hsu, Alexander J. Keeley, David G. Partridge, Shay Leary, et al. 2021. “Altered Subgenomic RNA Expression in SARS-CoV-2 B.1.1.7 Infections.” Cold Spring Harbor Laboratory. https://doi.org/10.1101/2021.03.02.433156.
Rambaut, Andrew, Edward C. Holmes, Áine O’Toole, Verity Hill, John T. McCrone, Christopher Ruis, Louis du Plessis, and Oliver G. Pybus. 2020. “A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology.” Nature Microbiology 5 (11): 1403–7.
Starr, Tyler N., Allison J. Greaney, Amin Addetia, William W. Hannon, Manish C. Choudhary, Adam S. Dingens, Jonathan Z. Li, and Jesse D. Bloom. 2021. “Prospective Mapping of Viral Mutations That Escape Antibodies Used to Treat COVID-19.” Science 371 (6531): 850–54.
Starr, Tyler N., Allison J. Greaney, Sarah K. Hilton, Daniel Ellis, Katharine H. D. Crawford, Adam S. Dingens, Mary Jane Navarro, et al. 2020. “Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding.” Cell 182 (5): 1295–1310.e20.
Suryadevara, Naveenchandra, Swathi Shrihari, Pavlo Gilchuk, Laura A. VanBlargan, Elad Binshtein, Seth J. Zost, Rachel S. Nargi, et al. 2021. “Neutralizing and Protective Human Monoclonal Antibodies Recognizing the N-Terminal Domain of the SARS-CoV-2 Spike Protein.” Cell 184 (9): 2316–31.e15.
Thomson, Emma C., Laura E. Rosen, James G. Shepherd, Roberto Spreafico, Ana da Silva Filipe, Jason A. Wojcechowskyj, Chris Davis, et al. 2021. “Circulating SARS-CoV-2 Spike N439K Variants Maintain Fitness While Evading Antibody-Mediated Immunity.” Cell 184 (5): 1171–87.e20.
Wang, Pengfei, Manoj S. Nair, Lihong Liu, Sho Iketani, Yang Luo, Yicheng Guo, Maple Wang, et al. 2021. “Antibody Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7.” Nature, March. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7 | Nature.
Acknowledgements
Sequencing of SARS-CoV-2 samples is undertaken by the Sheffield COVID-19 Genomics Group as part of the COG-UK CONSORTIUM and supported by funding from the Medical Research Council (MRC) part of UK Research & Innovation (UKRI), the National Institute of Health Research (NIHR) and Genome Research Limited, operating as the Wellcome Sanger Institute. MDP is funded by the NIHR Sheffield Biomedical Research Centre (BRC - IS-BRC-1215-20017). TIdS is supported by a Wellcome Trust Intermediate Clinical Fellowship (110058/Z/15/Z). SH is funded by The European & Developing Countries Clinical Trials Partnership. We thank all partners of and contributors to the COG-UK consortium, who are listed at https://www.cogconsortium.uk/about/ and here.
We acknowledge the authors from the originating laboratories responsible for obtaining the specimens and the submitting laboratories where genetic sequence data were generated and shared via the GISAID Initiative, on which some of this work is based. The complete information of the originating lab teams can be found here.

