[Updated information on the Genomic surveillance from Tocantins state, Brazil]
We report here the sequencing, assembling and the analyses of 78 new SARS-CoV-2 genomes from 22 cities in the Tocantins state. In order to construct a phylogenetic tree, we first aligned the 78 newly sequenced genomes and the reference genome as outgroup MAFFT v.7.480 [15]. The resulting alignment was subject to Maximum Likelihood phylogenetic analysis with IQ-TREE v.2.1.2 [16] under the Generalized Time Reversible GRT model of nucleotide substitution with empirical base frequencies (+F) and invariable sites (+I), as selected by the ModelFinder software. From the phylogeny, we observed two clearly distinguishable clusters composed of P.1 (also referred as Gamma lineage by the World Health Organization) and P.1.7 lineages (Figure 1).
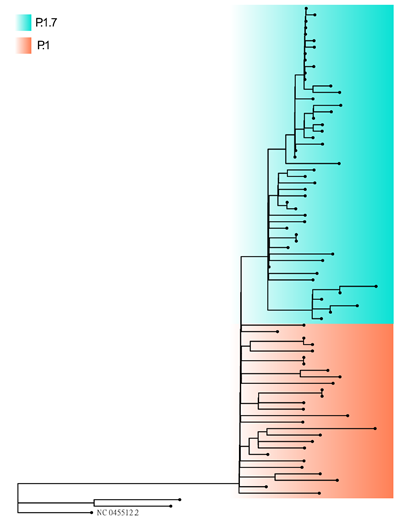
Figure 1. Phylogenetic tree of the 78 genomes newly assembled from Tocantins state. Sequences from the P.1.7 lineages are colored in blue, and the P.1 lineages are colored in orange. The maximum likelihood tree was built with IQ-TREE2.
A total of four Pango lineages were observed. The most frequently detected lineage group was P.1.7 (49 sequences or 62.8%), followed by P.1 (27 sequences or 34.6%) and B.1.1 and P.1, each with only one sequence. The P.1.7 lineage contains the P684H mutation on the spike gene, which was first observed on six of the 24 sequenced genomes that have been reported previously on our paper (Figure 2).
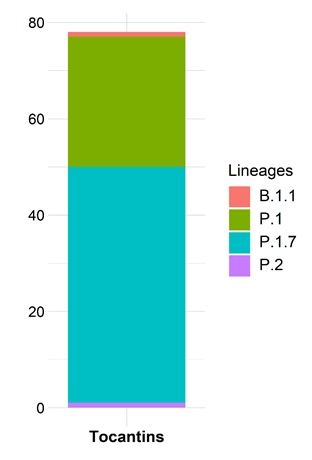
Figure 2. Lineages found on the 78 newly genomes sequenced from Tocantins states.
In addition, we identified 44 mutations of the SNPs type (Single Nucleotide Polymorphisms) and two mutations of the InDel type (Insertion-Deletion) in these genomes. Among the 44 SNPs identified, 42 are in coding regions of the viral genome and one in the 5’UTR region (C241T) and one in the 3’UTR region (C29499T). Moreover, 14 mutations were synonymous and 28 were missense variants, with 46.4% located at the spike gene (with 13 mutations: L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, P681H, T1027I, V1176F). The ORF1ab had a total of six non-synonymous mutations (S1188L, K1795Q, V2061F, P4715L, E5665D, T6566M). Six substitutions are in the N gene (P80R, S202C, S202T, R203K, G204R, Q409R). Two were found in the ORF3a (T223I and S253P) and one in ORF8 (E92K). The allele frequencies of the missense variants are reported in Figure 3.

Figure 3. Allele frequency (plotted as percentage) of missense SNPs found on the 78 SARS-CoV-2 genomes from Tocantins state.
Overall, now the Tocantins state has 230 SARS-CoV-2 genomes (108 sequenced by the Bioinformatics and Biotechnology Laboratory, Campus of Gurupi, Federal University of Tocantins in partnership with LACEN-Tocantins, 84 by the Adolfo Lutz Institute and 38 by the FIOCRUZ). The Genomic analysis with Pangolin reveals nine SARS-CoV-2 lineages circulating in Tocantins state. The most abundant lineage was P.1 accounting for 49.1% (113 genomes) of sequenced genomes. The second most common lineage was the P.1.7 (33.9% or 78 genomes), followed by the P.2 lineage (6.5% or 15 genomes) and B.1.1.28 (6.1% or 14 genomes) (Figure 4). We also point out that since its emergence and spread, the P.1 lineage has quickly become the dominant circulating in Tocantins state. We also observed a growth of the P.1.7 lineage in the last three months (Figure 5).
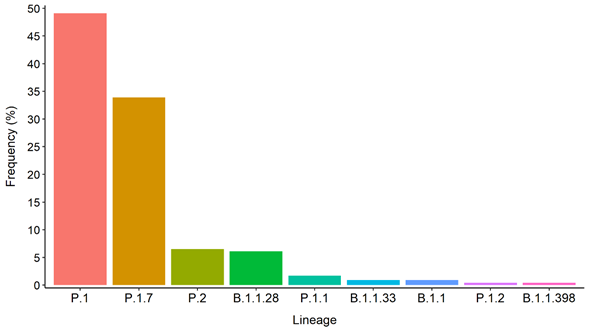
Figure 4. Distribution of the pangolin lineages found in Tocantins state.
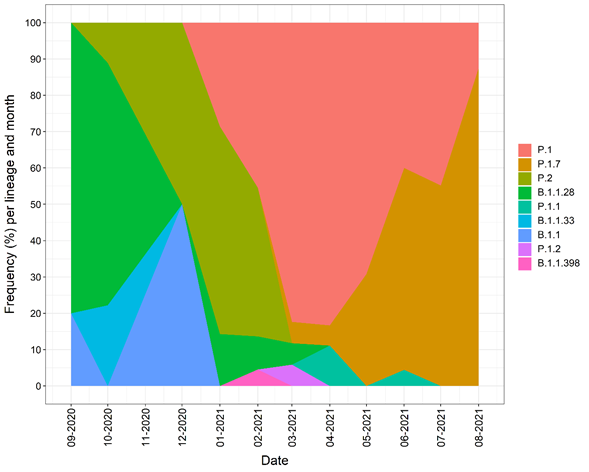
Figure 5. Frequency of the circulating lineages in Tocantins state.