Arnold W. Lambisia1, *, Joyce U. Nyiro1, John M. Morobe1, Timothy N. Makori1, Leonard Ndwiga1, Maureen W. Mburu1, Edidah O. Moraa1, Jennifer Musyoki1, Nickson Murunga1, Philip Bejon1,2, Lynette Isabella Ochola-Oyier1,2, D. James Nokes1,3, Charles N. Agoti 1,4 and George Githinji1,5, *
Affiliations.
- Kenya Medical Research Institute (KEMRI)-Wellcome Trust Research Programme (KWTRP), Kilifi, Kenya.
- Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
- School of Life Sciences and Zeeman Institute for Systems Biology and Infectious Disease Epidemiology Research (SBIDER), University of Warwick, Coventry, UK.
- School of Public Health, Pwani University, Kilifi, Kenya
- Department of Biochemistry and Biotechnology, Pwani University, Kilifi.
Corresponding author: [email protected]
Keywords: SARS-CoV-2, Kenya, Beta VOC, convergent evolution
Introduction
The Beta variant of concern was first detected on 26th June 2020 in South Africa and the first genomic detection was reported on 15th December 2020 in Kenya 1. In Kenya, the Beta VOC was in high transmission early in the third wave of SARS-CoV-2 infections and was replaced over time by the more transmissible Alpha and Delta variants 2–5. The last genomic report of Beta variant was from a case that was tested on 30th July 2021 from the Rift Valley region 1,3. Recently, we detected a Beta-like variant from a single sample collected at Kilifi County Hospital in coastal Kenya during routine surveillance. This report highlights the characteristics of this Beta variant that was collected on 29th September 2022. The participant had a history of fever, cough, and runny nose at the time of presenting to the health facility with no history of international travel two weeks before sample collection. The sample was collected at an outpatient clinic with no follow-ups post-discharge.
Methods
RNA extraction was carried out using QIAamp Viral RNA Mini Kit (Qiagen, Manchester, UK) and screening was carried out using TaqMan™ Fast Virus 1-Step Master Mix (Thermofisher, UK) as previously described 6. Reverse transcription was done using Lunascript (New England Biolabs, UK) and the cDNA amplified using Q5® Hot Start High-Fidelity 2x kit (New England Biolabs, UK) with V4.1 primers as previously described 7. Sequencing libraries were generated in duplicate using LSK-109 ligation kit for sequencing on ONT GridION platform and COVID-19 library kit (Illumina, USA) for sequencing on the Illumina Miseq. The consensus genome was generated from ONT reads (fastq and fast5) using the ARTIC bioinformatics protocol and the Dragen Lineage assignment application on BaseSpace for Illumina paired end reads. The genome was deposited on GISAID under accession number EPI_ISL_15654640.
Results
We observed a Beta-like sequence with additional six synonymous and 16 non-synonymous mutations within the ORF1ab, ORF3a, M, and S genes (Table 1 and Figure 1). The mutations were observed in the consensus genome from both the ONT and Illumina platforms. The additional mutations observed were predominantly seen within the Delta and Omicron VOC lineages and non-VOC lineages including B.1.1.456 and B.1.1.374 (Table 1). The additional alleles observed in the newly sequenced samples had sufficient read depth (>90%) compared to the Wuhan reference (Table 1 and Figure 2).

Figure 1: An alignment showing SNPs across SARS-CoV-2 genomes from the Beta-like (K290922) genome and other genomes from Alpha, Beta, Delta, Omicron and non-VOC variants relative to the Wuhan strain.
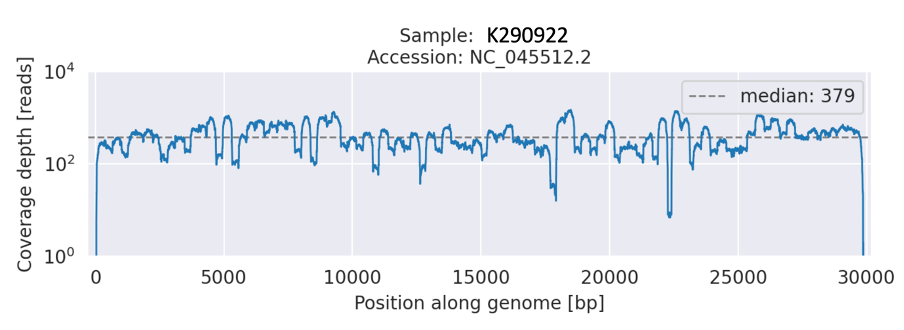
Figure 2: A line graph showing the read depth (Y-axis) across the genome (x-axis) of the Beta-like sequence.
Table 1: Description of additional mutations observed in the Beta-like S gene sequence
Amino acid Mutation Nucleotide mutation Total
Read depth Allele read depth Reference read depth Lineages/VOC where observed
S:G75A G21786C 231 227 4 Delta and Omicron
S:T95I C21846T 203 203 0 BA.1* (100%), AY.4.16 and AZ
S:E180K G22100A 1373 1317 56 B.1.1.374 (17%)
S:V213E G22200A 798 798 0 Omicron V213N/G BA.2
S:N764K C23854A 823 807 11 Omicron BA.4/5
S:I834V A24062G 304 302 2 B.1.1.456 (78%)
S:G75A, S:T95I, S:E180K and S:V213E mutations found within the N-terminal domain of the spike may affect the binding efficiency of the antibody thus influencing immune escape8.
Phylogenetic placement
The sequence clustered within B.1.351 sequences from Kenya (Figure 3). However, the new sequence contained additional mutations compared to previous sequenced Beta sequences from Kenya based on the genetic distance and time (Figure 3). Other global sequences with a similar genetic distance lacked the additional mutations observed in the K290922 sequence. Additionally**,** the Beta-like sequence clustered within Beta-like sequences in a maximum likelihood tree containing global sequences from other lineages (Figure 4).
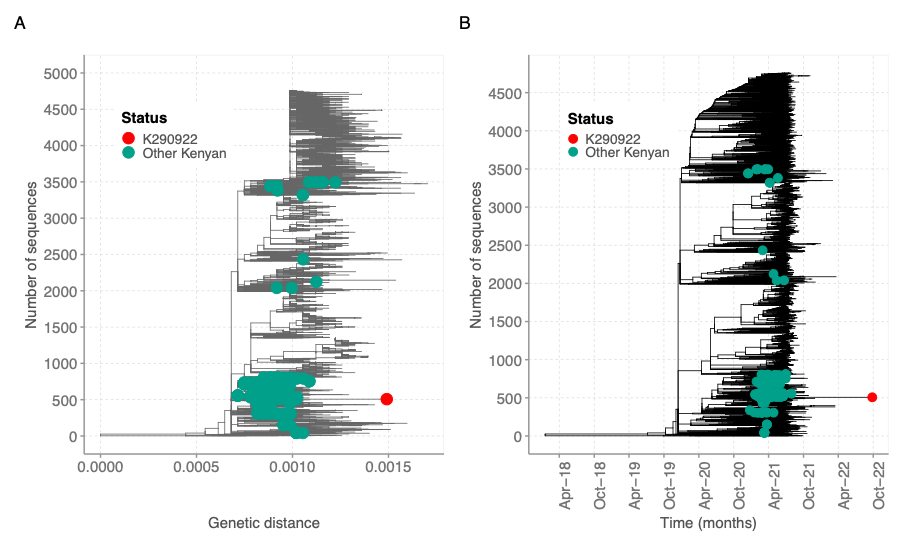
Figure 3: Genetic diversity of the beta-like sequence relative to a global reference dataset. A) A maximum likelihood tree showing the genetic diversity of the beta-like sequence (red dot) relative to 217 Kenyan Beta genomes (green dots) and a global reference set of 4500 genomes. B) A time resolved tree showing clustering of the beta-like genome to a global reference dataset as described earlier.
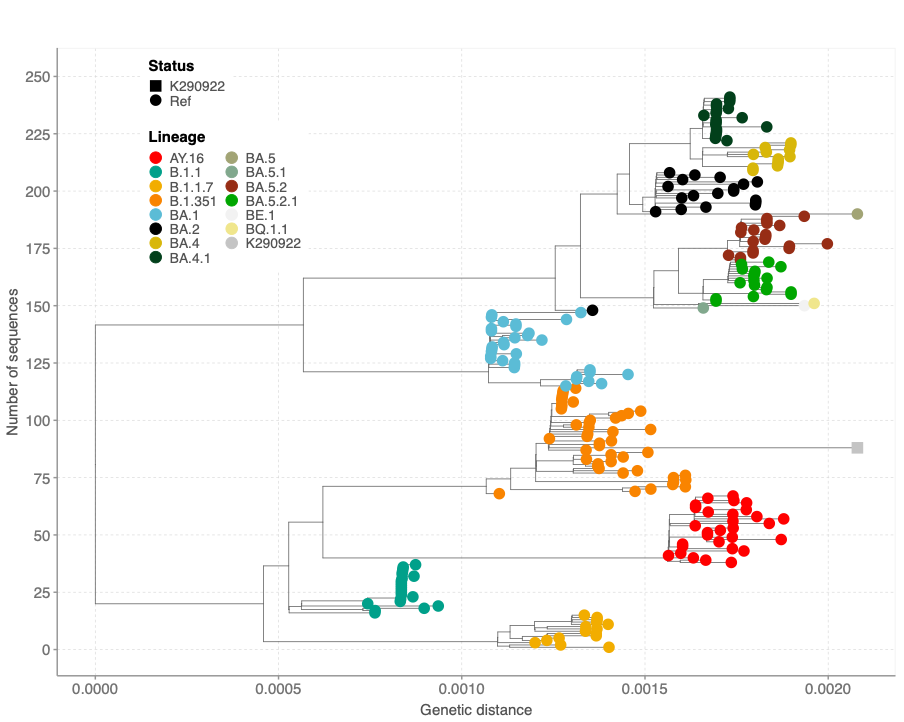
Figure 4: A maximum likelihood tree showing clustering of the Beta-like sequence (K290922) among other lineages that share mutations observed in the query sequence.
Discussion
We report identification a Beta VOC-like sequence in coastal Kenya with 22 additional mutations some of which have been identified in other VOCs. This finding suggests that some of the previous SARS-CoV-2 VOCs that have been undetected for months may be still in circulation in communities albeit at low frequencies. The declining testing rates further increases the chance of being missed by low numbers of routine surveillance. We speculate that this case may have been in recent contact with someone who had continued to shed the Beta VOC over a prolonged period. The additional non-synonymous mutations observed within the spike protein in this variant have been observed in other lineages suggest possible recombination (not detected in our analysis) or convergent evolution 9,10.
Funding statement :
This work is supported through grants from the National Institute for Health and Care Research (NIHR) (project references 17/63/82 and 16/136/33) using UK aid from the UK Government to support global health research, The UK Foreign, Commonwealth and Development Office and Wellcome Trust (grant# 220985/Z/20/Z) The views expressed in this publication are those of the author (s) and not necessarily those of NIHR or the Department of Health and Social Care, Foreign Commonwealth and Development Office.
References
-
Shu Y, McCauley J. GISAID: Global initiative on sharing all influenza data – from vision to reality. Eurosurveillance [Internet]. 2017;22(13):2–4. Available from: http://dx.doi.org/10.2807/1560-7917.ES.2017.22.13.30494
-
Campbell F, Archer B, Laurenson-Schafer H, Jinnai Y, Konings F, Batra N, et al. Increased transmissibility and global spread of SARSCoV- 2 variants of concern as at June 2021. Eurosurveillance [Internet]. 2021;26(24):1–6. Available from: http://dx.doi.org/10.2807/1560-7917.ES.2021.26.24.2100509
-
Kimita G, Nyataya J, Omuseni E, Sigei F, Lemtudo A, Muthanje E, et al. Temporal lineage replacements and dominance of imported variants of concern during the COVID-19 pandemic in Kenya. Commun Med. 2022;2(1):1–13.
-
Nasimiyu C, Matoke-muhia D, Rono GK, Osoro E, Daniel O. Imported SARS-COV-2 Variants of Concern Drove Spread of Infections Across Kenya During the Second Year of the Pandemic. 2022;1–31.
-
Earnest R, Uddin R, Matluk N, Park DJ, Lemieux JE, Grubaugh ND, et al. Article Comparative transmissibility of SARS-CoV-2 variants Delta and Alpha in New England , USA Graphical abstract Comparative transmissibility of SARS-CoV-2 variants Delta and Alpha in New England , USA. 2022;
-
Ochola-Oyier LI, Mohammed KS, de Laurent ZR, Omuoyo DO, Lewa C, Gicheru E, et al. An optimization of four SARS-CoV-2 qRT-PCR assays in a Kenyan laboratory to support the national COVID-19 rapid response teams. Wellcome Open Res. 2022;5:1–18.
-
Lambisia AW, Mohammed KS, Makori TO, Ndwiga L, Mburu MW, Morobe JM, et al. Optimization of the SARS-CoV-2 ARTIC Network V4 Primers and Whole Genome Sequencing Protocol. Front Med [Internet]. 2022 Feb 17;9(February):1–8. Available from: Frontiers | Optimization of the SARS-CoV-2 ARTIC Network V4 Primers and Whole Genome Sequencing Protocol
-
Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM, et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol [Internet]. 2021;19(7):409–24. Available from: SARS-CoV-2 variants, spike mutations and immune escape | Nature Reviews Microbiology
-
Shrestha LB, Foster C, Rawlinson W, Tedla N, Bull RA. Evolution of the SARS-CoV-2 omicron variants BA.1 to BA.5: Implications for immune escape and transmission. Rev Med Virol. 2022;32(5).
-
Focosi D. Convergent evolution within Omicron sub-variants [Internet]. 2022 [cited 2022 Oct 27]. Available from: https://twitter.com/dfocosi/status/1585175361180299265