Common Microdeletions in SARS-CoV-2 Sequences
Brianna Sierra Chrisman 1, Kelley Paskov 2, Nate Stockham 3, Jae-Yoon Jung 2, Maya Varma4, Peter Washington1, Dennis P. Wall 2,5
correspondance: [email protected] or [email protected]

[1] Department of Bioengineering, Stanford University
[2] Department of Biomedical Data Science, Stanford University
[3] Department of Neuroscience, Stanford University
[4] Department of Computer Science, Stanford University
[5] Department of Pediatrics (Systems Medicine), Stanford University
May 14, 2020
Summary
Researchers around the world are closely monitoring the mutational patterns of SARS-CoV-2, the virus that causes COVID-19 and which is the source of the current global pandemic. Most studies have used sequences available on the GISAID database 1 to do such analyses and have focused on single nucleotide polymorphisms. Less attention has been paid so far to small deletions or insertions, despite the known role of such structural variants in viral evolution.2,3
Here, we show 26 deletions in 15 different regions of the genome that are shared by at least 2 SARS-CoV-2 sequences. Most deletions are multiples of 3 bases long. Most interesting is a common ATG deletion at loci 1604, present in 386 sequences. There are also more than 30 independent and geographically diverse samples with various deletions between loci 507 and 517, a region rich in ATG subsequences. There are additional regional clusters of deletions.
With some of the raw reads that were used to construct the GISAID consensus sequences, we validate that these deletions are not obvious sequencing errors or a result of low-coverage regions. Interestingly, while most deletions are fairly homogeneous (around 90%), we occasionally see slight intrahost variation, with a small percentage of reads in a sample still retaining the reference sequence or having a different deletion nearby. Therefore, we hypothesize that these deletions may be near recombination hotspots. The deletions may either be inherited from SARS-CoV-2 viruses that recombined within a previous host or arise due to de novo recombination events.
Because we see evidence in the raw sequence data for interruption of a start codon in at least 1 paralog, and because of the complex architecture of the SARS-CoV-2 transcriptome4 we are curious about the impact of disrupting non-canonical start codons. We also emphasize how invaluable the raw or aligned reads are for better understanding of SARS-CoV-2 evolutionary dynamics. We urge the scientific community to make their raw reads publicly available if possible.
Results and Discussion
We found 192 unique deletions in the SARS-CoV-2 sequences available on GISAID, the locations of which are shown in Figure 1. The full set of deletions can be found at https://github.com/briannachrisman/SARS-CoV-2_Microdeletions/blob/master/deletions_full.csv. We focus the rest of our discussion and analysis on the non-singleton deletions, as we can be more confident that these deletions are not the result of a low-quality sample or sequencing error.
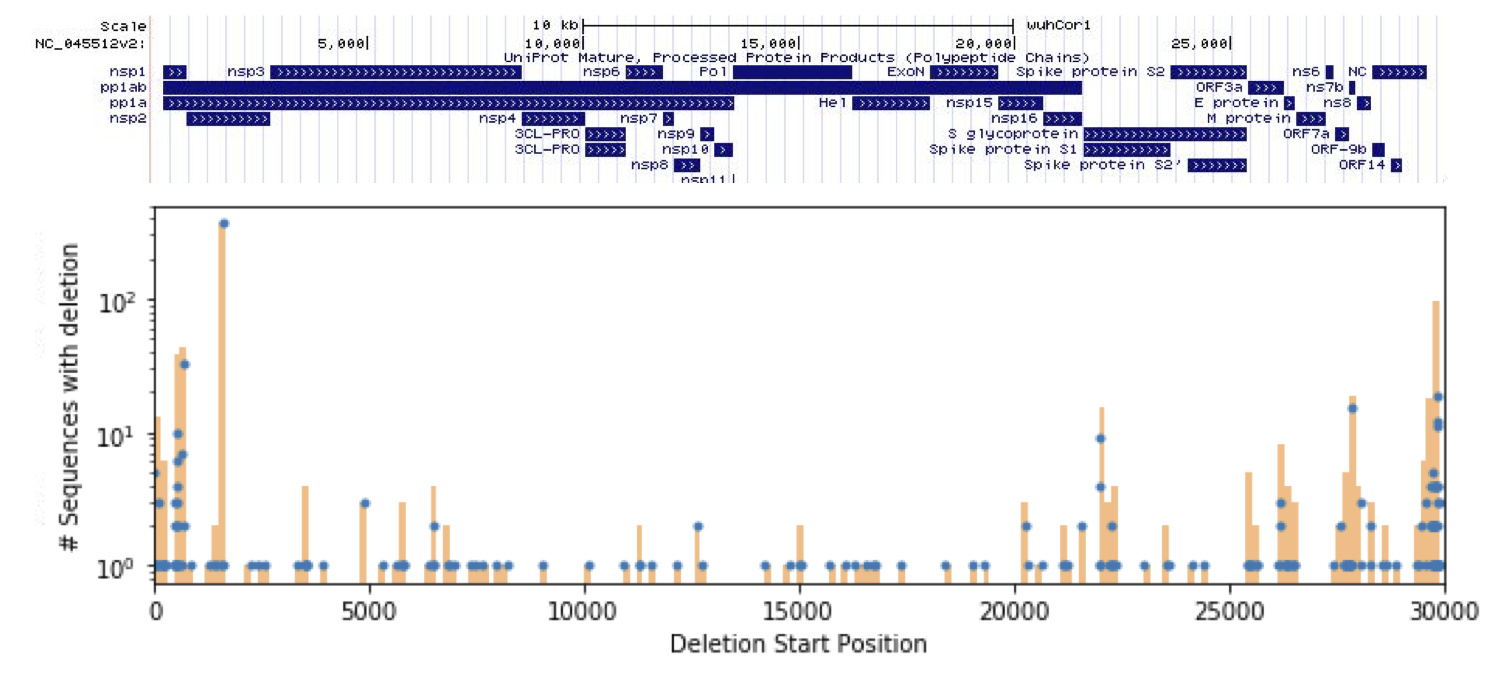
Figure 1: Deletions found in the GISIAD SARS-CoV-2 samples, overlayed with the UCSC genome browser annotations.
We find 26 non-singleton deletions, of which 21 are multiples of 3 bases long. These deletions would seemingly not cause frameshifts, supporting the hypothesis that these deletion calls are in fact biological, and not due to PCR or sequencing error. We describe the key features of these non-singleton deletions in Table 1.
Most interesting is site 1604, in which an ATG is deleted in 368 sequences. It is also highly homogeneous in the raw reads that we were able to access.
We also observe that many deletions occur at nearby sites (513, 515, and 517; 668 and 685; 21989 and 21990). While this could be due to low coverage or region-specific PCR and sequencing errors from the raw reads, this does not seem to be the case. These regions do not have low coverage, and we do not see high rates of heterogeneity in and around these regions within samples, which has commonly been used to flag other mutations suspected to be the result of sequencing error 5 or hypermutability.6
While the heterogeneity does not seem high enough to suspect sequencing error, most deletions for which we were able to retain raw reads were not completely homogeneous – many samples are 80-90% homogeneous for a given deletion. Many samples still retain reads without a deletion or have a slightly different deletion nearby. We therefore hypothesize that these regions may be recombination hotspots. The deletions would therefore be artifacts of SARS-CoV-2 recombination, either when the virus was in a previous host or de novo recombination events in the sample’s host. These patterns can be observed in Figures 3,5,7,9.
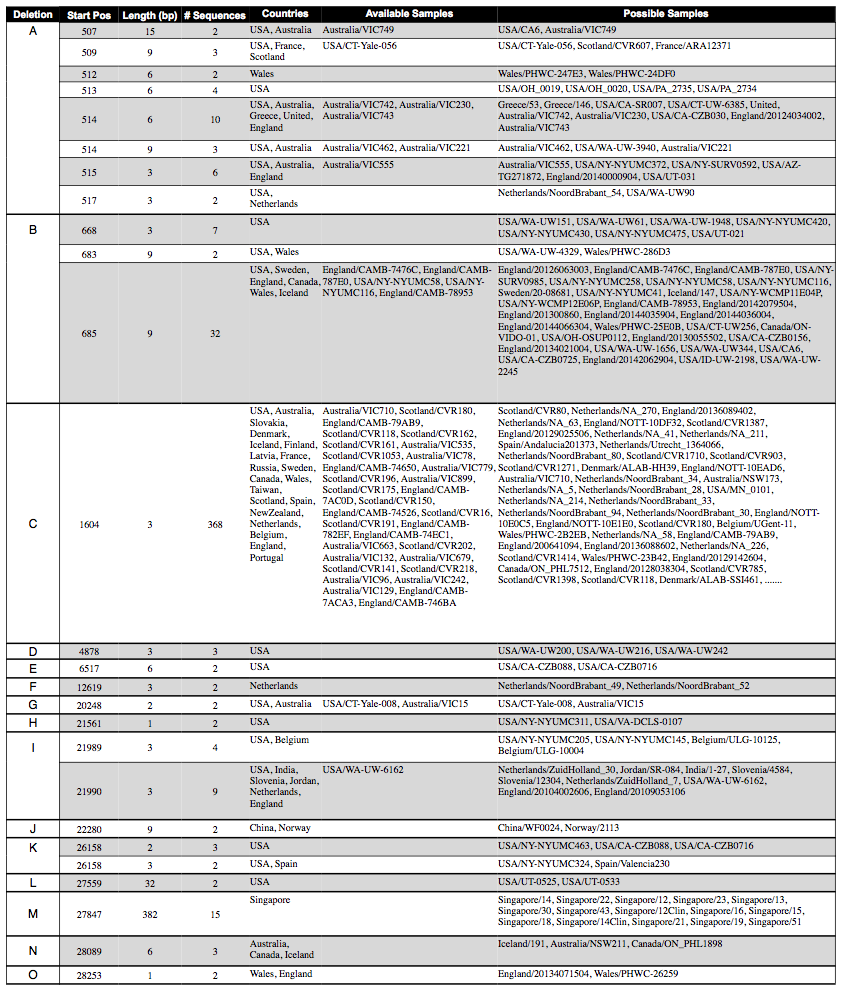
Table 1: Deletions that were found in at least two of the SARS-CoV-2 sequences on GISAID, categorized into regions A-O based on genomic location. We note the sequences in which the raw reads were also available to further validate the deletion.
We find the 3-basepair deletion at 1604 particularly interesting. This deletion is of start codon (ATG) in one of the non-canonical reading frames. Given that SARS-CoV-2 expresses many non-canonical transcripts 4, it is possible that this deletion prevents expression of a non-canonical transcript. Important questions remain about reliability of the consensus sequences and the stability of phylogenetic trees built from GISAID data. 7 However, once answered, future work using phylogenetic approaches for analysis on parallel evolution and adaptive selection should help to characterize the potential mechanisms for this non-canonical deletion.
Deletion Region A: 507-517
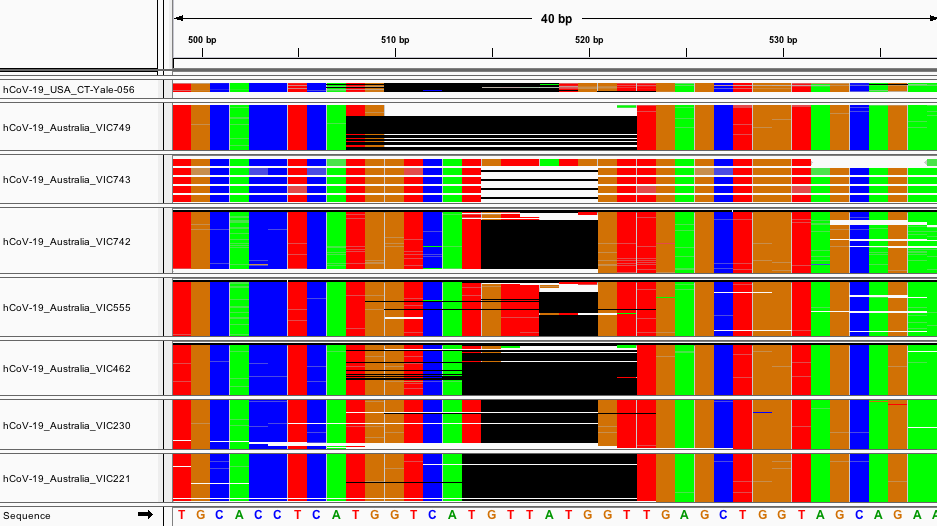
Fig. 2: Multiple Sequence Alignment of several sequences with deletion A.
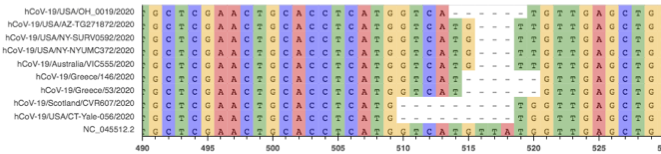
Fig 3: Aligned raw reads from available samples with deletion A.
There are several small deletions of various lengths between 507-517. Notably, all deletions have lengths that are multiples of 3 bps. Also interesting is that this region is heavily enriched for non-canonical start codons. In the 14 bases from 507-520, there are 3 start codons in non-canonical reading frames. All of the deletions in set A disrupt a start codon. MSA and raw read alignment are shown in Fig. 2. Several variations of the deletions shown would be equivalent via local realignment.
Deletion Region B: 668-685
We found 21 non-singleton deletions between 668 and 685. All mutations were multiples of 3 bps long. We see from the raw reads that these deletions have a small degree of heterogeneity - in several reads we see the 9bp mutation at 685 is replaced by a similar deletion several bases shifted. However, it should be noted that the COG- samples from England are of lower quality and coverage than the reads provided by other sequencing centers, so it is possible the heterogeneity is an artifact.

Fig. 4: MSA of several sequences with deletion B
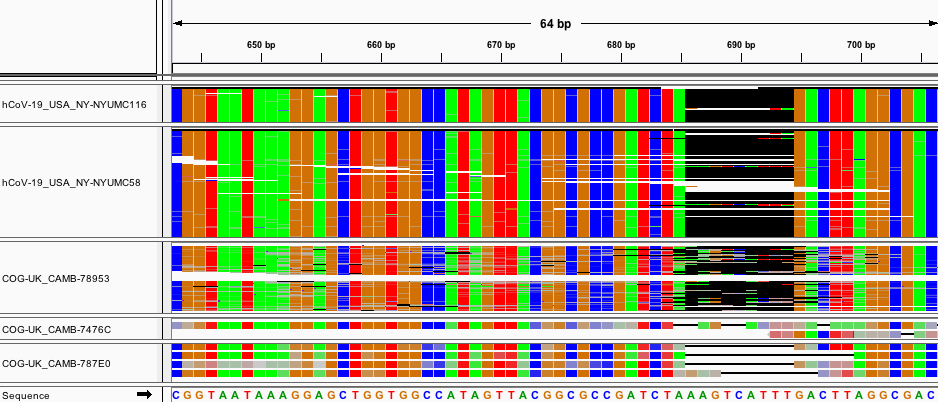
Fig. 5: Aligned raw reads from available samples with deletion B.
Deletion Region C: 1604
368 samples contained a 3 bp deletion at loci 1604. These samples came from all around the world and were sequenced using both Oxford Nanopore and Illumina platforms. We performed an alignment for several samples that were publicly available. After quality filtering, adaptor trimming, and PCR duplicate removal, the deletion persists in the alignment at a high homogeneity. We see no evidence that this deletion is an artifact of sequencing error.
Interestingly, this deletion does not exhibit the same properties as the other clusters of deletions. There are not neighboring deletions (as in deletion region A) and the raw read alignments have nearly 0% heterogeneity. In this way, it seems less plausible that this deletion is related to a nearby recombination hotspot.
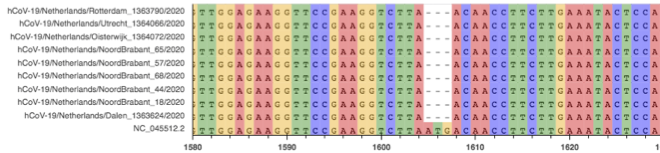
Fig. 6: MSA of several sequences with deletion C
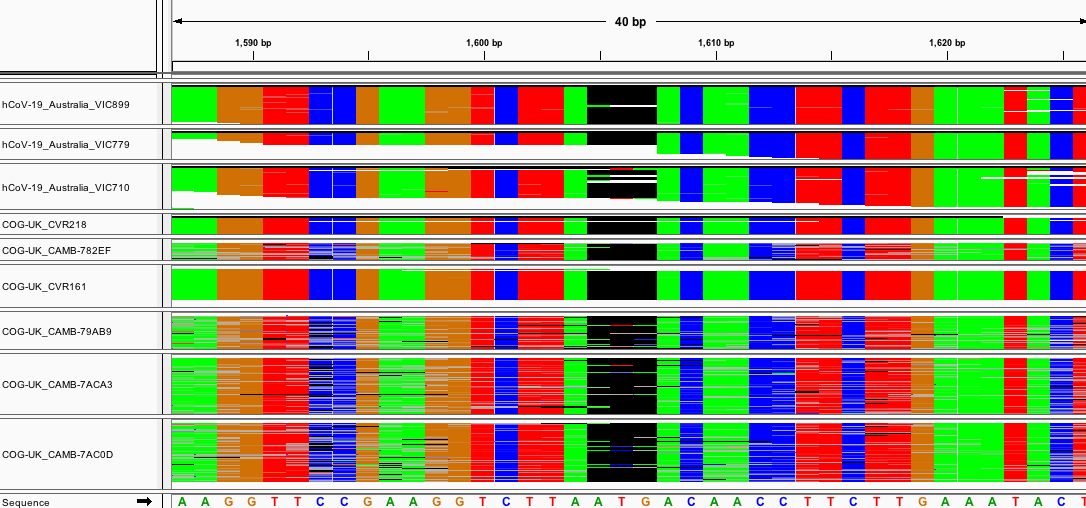
Fig. 7: Aligned raw reads from several of the available samples with deletion C.
Deletion Region I: 21989-21990
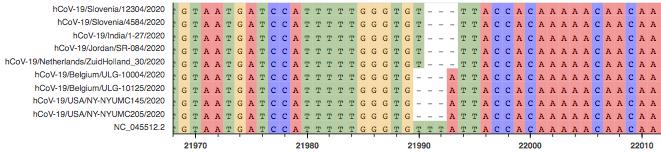
Fig. 8: MSA of several sequences with deletion I
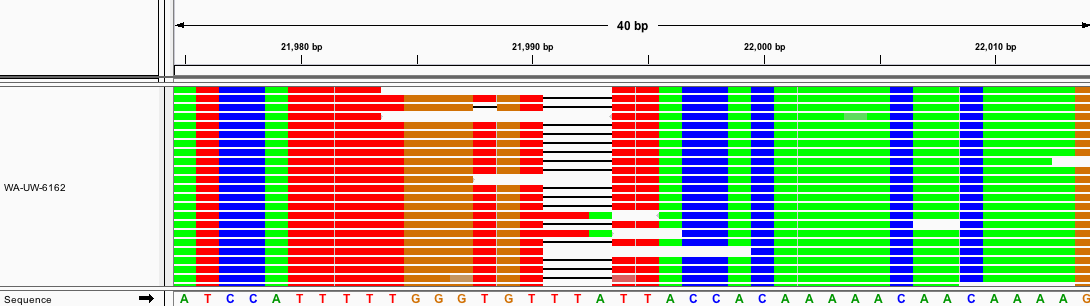
Fig. 9: Aligned raw reads from available samples with deletion I.
Other Deletion Regions
We briefly describe the rest of the deletion regions, which we were unable to validate, as no raw reads containing the deletion were available. Many sequences have low quality or unknown calls flanking the deletion region. It is possible that the low quality of the calls comes from high heterogeneity in surrounding sites and this high heterogeneity is due to de novo recombination. This is speculation, and needs to be validated with raw reads.
The two samples that contained the 3-basepair deletion at loci 12619 were from the same lab in the Netherlands, and were also both sequenced with the Oxford Nanopore platform. It is possible this deletion is merely a sequencing error. After filtering for quality and PCR duplicates, the reads containing deletion G were both low read depth (<10). We cannot be certain whether this deletion is merely a sequencing or PCR artifact, although the two samples were sequenced on different platforms (Oxford Nanopore and Illumina). Deletion N is a 382-nucleotide deletion in the spike protein. Although none of the raw reads of the sequences with the deletion were available, this deletion has been observed and characterized in previous literature.8
Methods
Data Access
We accessed all GISAID SARS-CoV-2 sequences on May 7, 2020. We filtered to high coverage reads encompassing the entire SARS-CoV-2 genome (>=29000 bps), leaving 12,403 sequences.
Multiple Sequence Alignment
We used the augur pipeline provided from Nextstrain 9 to align sequences and build maximum likelihood trees. We used MAFFT10 to perform multiple sequence alignment with NC_045512.2 as the reference sequence. We locally realigned the two deletion calls that were equivalent using python.
Raw Read Alignment
We downloaded the fastq files for the raw reads using the NCBI SRA Run browser. NYU Langone Health provided us with the aligned reads for USA/NY-NYUMC116 and and USA/NY-NYUMC58.11
We quality filtered the reads using fastp for Illumina reads, with a qualified quality phred cutoff of 20, an unqualified percent limit of 20, and a required length of 50. We used Nanofilt for Oxford Nanopore reads, with a minimum quality of 10, a minimum length of 500, and a headcrop of 50. We used bwa-mem to align illumina reads to the reference genome, following the standard paired or single end read pipelines as appropriate. We used minimap2 to align the oxford nanopore reads to the reference genome. We marked and removed PCR duplicates using GATK’s MarkDuplicates. We used lofreq to quality score the indels and perform local realignment.
Acknowledgements
We thank all of the labs that submitted their sequences to the GISAID database. The full acknowledgement table can be found at https://github.com/briannachrisman/SARS-CoV-2_Microdeletions/blob/master/acknowledgments.pdf. We thank the public health laboratories VIDRL and MDU-PHL at The Peter Doherty Institute for Infection and Immunity for providing over 1000 high quality raw reads to NCBI. Thank you NYU Langone SARS-CoV2 Sequencing Team’s Matthew T Maurano, Matija Snuderl, and Adriana Heguy for providing many of their raw reads.
References
[1] Yuelong Shu and John McCauley. “GISAID: Global initiative on sharing all influenza data–from vision to reality”. Eurosurveillance 22.13(2017).9
[2] Natasha Wood et al. “HIV evolution in early infection: selection pressures, patterns of insertion and deletion, and the impact of APOBEC”. *PLoSpathogens& 5.5 (2009).
[3] Jonathan A McCullers et al. “Reassortment and insertion-deletion arestrategies for the evolution of influenza B viruses in nature”. Journalof virology 73.9 (1999), pp. 7343–7348.
[4] Dongwan Kim et al. “The architecture of SARS-CoV-2 transcriptome”.Cell(2020).
[5] Heng Li. “Toward better understanding of artifacts in variant calling from high-coverage samples”. Bioinformatics 30.20 (2014), pp. 2843–2851.
[6] Esteban Domingo, Julie Sheldon, and Celia Perales. “Viral quasispecies evolution”. Microbiol. Mol. Biol. Rev. 76.2 (2012), pp. 159–216.
[7] Nicola De Maio et al. “Issues with SARS-CoV-2 sequencing data”. Virological.org (Accessed May 7, 2020).
[8] Yvonne Su et al. “Discovery of a 382-nt deletion during the early evolution of SARS-CoV-2”. bioRxiv(2020).
[9] Richard A Neher and Trevor Bedford. “Nextflu: real-time tracking ofseasonal influenza virus evolution in humans”. Bioinformatics 31.21(2015), pp. 3546–3548.
[10] Kazutaka Katoh, George Asimenos, and Hiroyuki Toh. “Multiple align-ment of DNA sequences with MAFFT”. In:Bioinformatics for DNA sequence analysis. Springer, 2009, pp. 39–64.
[11] Matthew T Maurano et al. “Sequencing identifies multiple, early introductions of SARS-CoV2 to New York City Region”. medRxiv (2020).