B.1.258∆, a SARS-CoV-2 variant with ∆H69/∆V70 in the Spike protein circulating in the Czech Republic and Slovakia
Broňa Brejová1,* , Viktória Hodorová2, Kristína Boršová3,4, Viktória Čabanová3, Lenka Reizigová5,6, Evan D. Paul7, Pavol Čekan7, Boris Klempa3, Jozef Nosek2, Tomáš Vinař1,*
Affiliations:
1 Faculty of Mathematics, Physics and Informatics, Comenius University in Bratislava, Mlynská dolina, 842 48 Bratislava, Slovak Republic
2 Department of Biochemistry, Faculty of Natural Sciences, Comenius University in Bratislava, Ilkovičova 6, 842 15 Bratislava, Slovak Republic
3 Institute of Virology, Biomedical Research Center of the Slovak Academy of Sciences, Dúbravská cesta 9, 845 05 Bratislava, Slovak Republic
4 Department of Microbiology and Virology, Faculty of Natural Sciences, Comenius University in Bratislava, Ilkovičova 6, 842 15 Bratislava, Slovak Republic
5 Regional Authority of Public Health, Trenčín, Slovakia
6 Department of Laboratory Medicine, Faculty of Healthcare and Social Work, Trnava University, Trnava, Slovakia
7 MultiplexDX, Bratislava, Slovakia
*Address for correspondence:
Faculty of Mathematics, Physics and Informatics, Comenius University in Bratislava, Mlynská dolina, 842 48 Bratislava, Slovak Republic; [email protected]; [email protected]
Summary
SARS-CoV-2 mutants carrying the ∆H69/∆V70 deletion in the amino terminal domain of the Spike protein emerged independently in at least six lineages of the virus (namely, B.1.1.7, B.1.1.298, B.1.160, B.1.177, B.1.258, B.1.375). Routine RT-qPCR tests including TaqPath or similar assays based on a drop-out of the Spike gene target are incapable of distinguishing among these lineages and often lead to the false conclusion that clinical samples contain the B.1.1.7 variant, which recently emerged in the United Kingdom and is quickly spreading through the human population. We analyzed SARS-CoV-2 samples collected from various regions of Slovakia between November and December 2020 that were presumed to contain the B.1.1.7 variant due to traveling history of the virus carriers or their contacts. Sequencing of these isolates revealed that although in some cases the samples were indeed confirmed as B.1.1.7, a substantial fraction of isolates contained another ∆H69/∆V70 carrying mutant belonging to the lineage B.1.258, which has been circulating in Central Europe since August 2020, long before the import of B.1.1.7. Phylogenetic analysis shows that the early sublineage of B.1.258 acquired the N439K substitution in the receptor binding domain (RBD) of the Spike protein and, later on, also the deletion ∆H69/∆V70 in the Spike N-terminal domain (NTD). This variant is particularly common in several European countries including Czech Republic and Slovakia, and we propose to name it B.1.258∆.
Introduction
Monitoring clinical samples by rapid and near real-time genome sequencing of the SARS-CoV-2 virus provides an important means for policy decisions in combating the COVID-19 pandemic (Oude Munnink et al. 2020). In particular, the emergence of new virus variants exhibiting enhanced infectivity such as B.1.1.7 (20I/501Y.V1), B.1.351 (20H/501Y.V2), and P.1 (20J/501Y.V3) recently detected in the United Kingdom, South Africa, and Brazil, respectively, increased the attention of national public health authorities (Rambaut et al. 2020a; Tegally et al. 2020; Kosakovsky Pond et al. 2020; NIoID 2021; Faria et al. 2021; Naveca et al. 2021a,b).
Here, we describe a variant named B.1.258∆ carrying the ∆H69/∆V70 mutation in the Spike protein that has emerged within the B.1.258 clade and has been prevalent in the Czech Republic (approx. 59% out of 251 sequenced samples between September and December 2020), Slovakia (25% out of 72 sequenced samples in the same months), and several other countries (Table 1). The ∆H69/∆V70 deletion in the Spike N-terminal domain (NTD) is associated with increased infectivity and evasion of the immune response (Kemp et al. 2020a) and evidence suggests that this mutation has arisen in B.1.258 independently of the B.1.1.7 variant. The deletion is likely to cause a drop-out of the Spike gene target in TaqPath RT-qPCR (Volz et al. 2020; Washington et al. 2020; Bal et al. 2021; Borges et al. 2021) and other assays targeting this deletion, and thus its carriers can be easily misidentified as B.1.1.7 (as it recently happened in Slovakia, see PHAoSR, 2021).
The B.1.258∆ variant also contains the N439K mutation in the receptor binding domain (RBD) of the Spike protein, likely preceding the emergence of the ∆H69/∆V70 mutation in this lineage. The N439K substitution enhances the binding affinity to the angiotensin-converting enzyme 2 (ACE2) receptor and has been shown to facilitate immune escape from a panel of neutralizing monoclonal antibodies, as well as from polyclonal sera from persons recovered from the infection. This mutation is also associated with slightly higher viral loads (Thomson 2021). The deletion ∆H69/∆V70 emerged recurrently in diverse lineages and it frequently co-occurs with mutations in the Spike RBD such as N439K, Y453F, and N501Y (Kemp et al. 2020b; Lassaunière et al. 2020; McCarthy et al. 2020; Rambaut et al. 2020; Bazykin et al. 2021; Larsen et al. 2021). It has been demonstrated that ∆H69/∆V70 enhances the virus infectivity by two-folds in a pseudotyping assay and also compensates for mutations in the Spike RBD that lower the infectivity (e.g. N501Y). In addition, a non-RBD binding monoclonal antibody is less potent against the ∆H69/∆V70 mutant (Kemp et al. 2020a). Other non-synonymous mutations characteristic for most of the B.1.258∆ samples result in amino acid substitutions in proteins involved in virus replication, i.e. M101I, localized in the dimerization motif ‘GXXXG’ (100-104) of RNA-binding protein NSP9, V720I in the palm subdomain of RNA-dependent RNA polymerase NSP12, and A598S near the C-terminus of helicase NSP13 (Zhang et al. 2020; Wang et al. 2020).
The association of mutations in the B.1.258∆ variant with increased virulence and evasion of immune responses taken together with the high prevalence in several countries (notably the Czech Republic, currently reporting one of the highest incidences of new cases per 100,000 population in the world) warrants further investigation of this variant.
Results and Discussion
The ∆H69/∆V70 mutation has arisen independently at least six times in different SARS-CoV-2 lineages (Figure 1). Besides the B.1.258∆ and B.1.1.7 variants, the mutation has also been observed in B.1.1.298 (Denmark outbreak associated with mink farms; Lassaunière et al. 2020; Larsen et al. 2021) and B.1.375 (a clade originating in the United States; Larsen and Worobey, 2020; Moreno et al. 2021). Interestingly, the ∆H69/∆V70 mutation has recently emerged within previously characterized clades EU1 (B.1.177) and EU2 (B.1.160). This recurrent emergence even within well-established clades supports the hypothesis that ∆H69/∆V70 mutation can compensate for other mutations that would by themselves lower the infectivity and in concert with those other mutations can lead to new variants with increased fitness and potential to evade the immune system response (Kemp et al. 2020b).
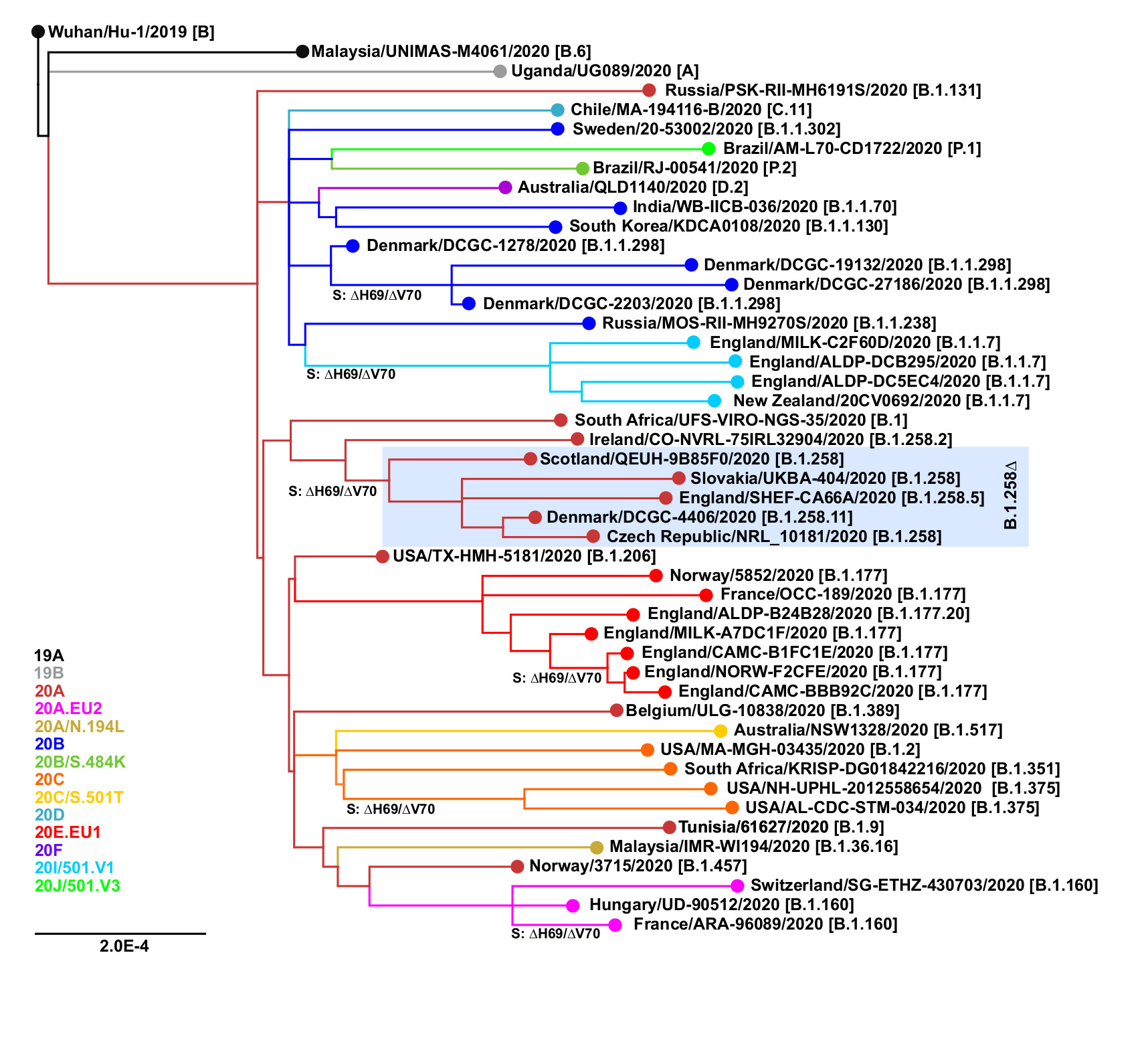
Figure 1. Recurrent emergence of the ∆H69/∆V70 mutation. The points of emergence of the ∆H69/∆V70 deletion are marked. Nextclade (https://clades.nextstrain.org; Hadfield et al. 2018) assignments are shown in color. Pangolin lineages (https://pangolin.cog-uk.io; Rambaut et al., 2020b) are indicated in brackets.
The earliest samples of the B.1.258∆ variant have been observed in Switzerland and in the United Kingdom at the beginning of August 2020 (Figure 2). The outgroup sample that already contains the Spike N439K mutation, but not the ∆H69/∆V70 deletion, has been sampled in Romania as early as May 13, 2020; before that, the outgroup that does not contain N439K has been observed in England on March 22, 2020 (EPI_ISL_423656). Subsequently, the B.1.258∆ variant has spread and gained significant prevalence in multiple countries, including the Czech Republic, Sweden, Slovakia, Poland, Denmark, and Austria (Table 1). Note that clade B.1.258 also includes sequences without the ∆H69/∆V70 deletion. In particular, most samples in sublineages B.1.258.1-B.1.258.3 and B.1.258.14-B.1.258.16 do not have the deletion, while those in B.1.258.4-B.1.258.12 and B.1.258.17-B.1.258.21 mostly have it.
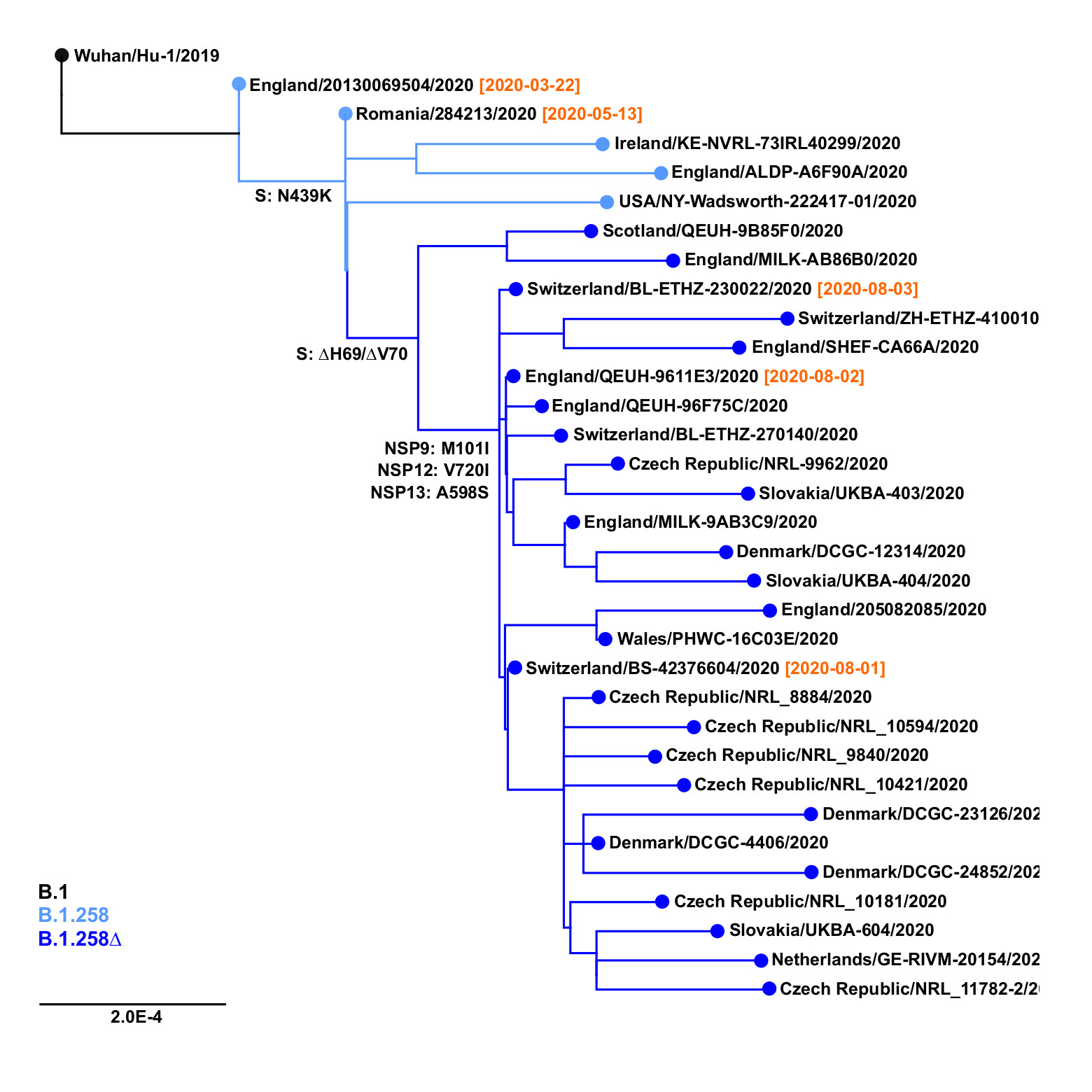
Figure 2. Origins of B.1.258∆ variant. Positions of mutations S:N439K, S:∆H69/∆V70 and additional substitutions defining the variant (i.e. NSP9:M101I, NSP12:V720I, NSP13:A598S) are marked. Collection dates are shown in samples nearest to the important branching points.
Table 1. Prevalence of B.1.258∆ and B.1.1.7 variants in selected countries between September and December 2020. The numbers represent the percentage of all samples uploaded to GISAID within the period. Only percentages based on at least 20 samples are shown, numbers based on fewer than 100 samples are shown in parentheses.
| B.1.258∆ | B.1.1.7 | |||||||
|
2020-09 |
2020-10 |
2020-11 |
2020-12 |
2020-09 |
2020-10 |
2020-11 |
2020-12 |
|
| Austria |
(0) |
22 |
(0) |
0 |
||||
| Czech Republic |
(40) |
66 |
(54) |
(0) |
0 |
(0) |
||
| Denmark |
11 |
16 |
10 |
9 |
0 |
0 |
<1 |
1 |
| Germany |
6 |
7 |
9 |
11 |
0 |
0 |
<1 |
3 |
| Iceland |
2 |
5 |
8 |
4 |
0 |
0 |
0 |
5 |
| Italy |
0 |
0 |
<1 |
2 |
0 |
0 |
0 |
14 |
| Netherlands |
<1 |
4 |
3 |
8 |
0 |
0 |
<1 |
7 |
| Poland |
29 |
(23) |
0 |
(1) |
||||
| Slovakia |
(19) |
(38) |
(0) |
(41) |
||||
| Sweden |
(2) |
(1) |
(2) |
43 |
(0) |
(0) |
(0) |
5 |
| Switzerland |
2 |
2 |
2 |
4 |
0 |
0 |
<1 |
2 |
| USA |
<1 |
0 |
0 |
<1 |
0 |
0 |
0 |
1 |
| United Kingdom |
3 |
3 |
2 |
1 |
<1 |
<1 |
6 |
43 |
In addition to sequence analyses, we used a recently developed RT-qPCR assay which permits differentiation of the B.1.1.7 variant from other variants harboring only the ΔH69/ΔV70 deletion (Kovacova et al. 2021) in order to analyze a panel of 122 SARS-CoV-2 positive clinical samples collected within a mass testing campaign in the city of Trenčín (Western Slovakia) in December 2020. The assay indicated that 50/122 (41.0%) of the samples were carrying only the ∆H69/∆V70 deletion while only 5/122 (4.1%) were identified as the B.1.1.7 lineage. Interestingly, analysis of the Ct values from the routine RT-qPCR assay showed significantly lower Ct values in the swab samples for both B.1.1.7 and B.1.258∆ samples, reflecting significantly higher viral loads in patients carrying these variants (Figure 3).
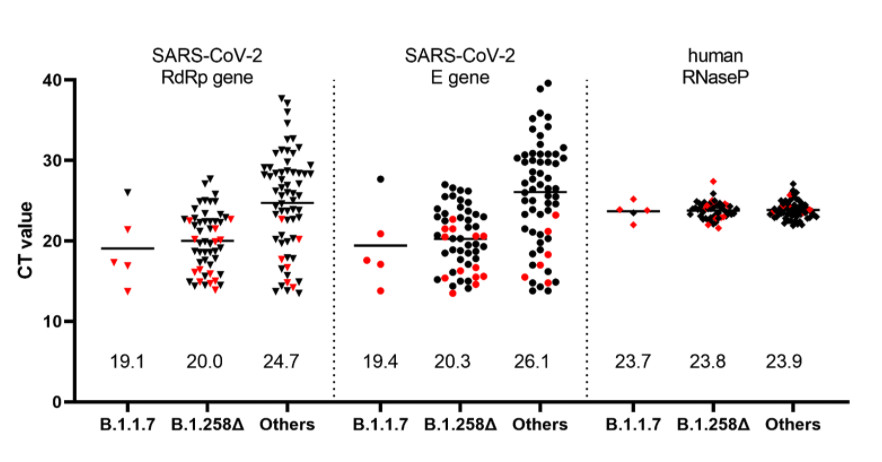
Figure 3. Ct values in the swab specimens collected in December 2020 in the city of Trenčín (Western Slovakia) grouped according to the identified SARS-CoV-2 lineages. The samples were initially sorted into the three groups by the RT-qPCR assay differentiating B.1.1.7 from other variants carrying ∆H69/∆V70 deletion (Kovacova et al. 2021). The presented values are the outcome of routine SARS-CoV-2 RT-qPCR assay targeting RdRp, E, and human RNase P genes. Human RNase P assay was used as a control and indicates that the observed Ct differences are unaffected by the quality of the sample collection and RNA extraction. Proper classification of data points marked in red was further confirmed by sequencing. Numerical values shown above the x-axis indicate the mean values.
Methods
The GISAID database was downloaded on February 3, 2021. The mutations in individual sequences were identified by comparison to the Wuhan/Hu-1/2019 sample (NCBI ID: NC_045512.2), and variant labels were assigned based on characteristic mutations identified by manual examination of the NextClade tree (for B.1.258∆, 4 out of 5 mutations G12988T G15598A G18028T T24910C T26972C were required; for B.1.1.7 variant, 11 out of 13 mutations C3267T C5388A T6954C A23063T C23271A C23604A C23709T T24506G G24914C C27972T G28048T A28111G C28977T). The samples shown in the phylogenetic trees were selected manually to cover significant lineages of interest (B.1.258∆ variant in different countries and its outgroups, lineages containing ∆H69/∆V70 mutation and their outgroups). Phylogenetic trees were built using Augur pipeline v. 6 (Hadfield et al. 2018). The prevalence was assessed based on all samples in GISAID for a particular country carrying particular characteristic mutations with collection dates in a particular month.
RT-qPCR assays were performed on RNA extracted by the Biomek i5 Automated Workstation using the RNAdvanced Viral kit (Beckman Coulter, Indianapolis, Indiana, USA) from swab samples collected within routine SARS-CoV-2 diagnostics (the samples were provided to researchers by health authorities without any personal identification of patients). Besides rTEST COVID-19 RT-qPCR Allplex kit (MultiplexDX, Bratislava, Slovakia) targeting the RNA-dependent RNA polymerase (RdRp) and Envelope (E) genes, the newly developed rTEST COVID-19 qPCR B.1.1.7 kit (MultiplexDX, Bratislava, Slovakia) was used to differentiate B.1.1.7 and B.1.258∆ variants (Kovacova et al. 2021). The real time PCR was performed on a QuantStudio™ 5 Real-Time PCR System (Applied Biosystem, Foster City, California, USA).
The genome sequences of SARS-CoV-2 isolates were determined on a MinION sequencer (Oxford Nanopore Technologies) using a protocol based on PCR-tiling of 2-kb long amplicons (Resende et al. 2020).
Conclusions
We have described a B.1.258∆ variant of the SARS-CoV-2 virus that contains the S:N439K mutation shown to enhance the binding affinity of the Spike protein to human ACE2 receptor and facilitating escape from immune response, the S:∆H69/∆V70 mutation, which is known to increase viral infectivity, as well as several other non-synonymous mutations in NSP9, NSP12 and NSP13 likely affecting viral replication. RT-qPCR analysis on random samples collected during a mass testing campaign indicate that B.1.258∆ samples carry higher viral loads compared to other strains, similarly as in the case of the B.1.1.7 variant.
Interestingly, the B.1.258.17 sublineage of B.1.258∆ has accumulated a higher number of mutations compared to other B.1.258∆ samples, including additional substitutions in the Spike protein (L189F, V772I) and helicase NSP13 (P53L). It also has a substitution Q185H in the second B cell epitope of ORF3a protein involved in apoptosis induction (Ren et al. 2020).
Our characterization of a novel B.1.258∆ variant that has emerged in several European countries and appears to result in higher viral loads highlights the importance of vigilant genomic surveillance methods in properly identifying and tracking SARS-CoV-2 variants that display the potential to derail worldwide efforts to mitigate the pandemic.
Acknowledgements. We gratefully acknowledge the authors from the originating laboratories responsible for obtaining the specimens, as well as the submitting laboratories where the genome data were generated and shared via GISAID (https://www.gisaid.org/), on which this research is based GISAID Acknowledgements.pdf (51.3 KB)
Funding. Our research was supported by grants from the Slovak Research and Development Agency (APVV-18-0239 to JN, PP-COVID-20-0017 to BK, and PP-COVID-20-0116 to PC and BK), the Scientific Grant Agency (VEGA 1/0463/20 to BB, VEGA 1/0458/18 to TV), and the European Union’s Horizon 2020 research and innovation program (EVA-GLOBAL project #871029 to BK and PANGAIA project #872539). The research was also supported in part by OPII project ITMS2014: 313011ATL7.
References
- Bal, A., et al. (2020) Two-step strategy for the identification of SARS-CoV-2 variants co-occurring with spike deletion H69-V70, Lyon, France, August to December 2020. medRxiv [Preprint]. doi:10.1101/2020.11.10.20228528
- Bazykin, G.A., et al. (2021) Emergence of Y453F and Δ69-70HV mutations in a lymphoma patient with long-term COVID-19. virological.org
- Borges, V., et al. (2021) Tracking SARS-CoV-2 VOC 202012/01 (lineage B.1.1.7) dissemination in Portugal: insights from nationwide RT-PCR Spike gene drop out data. virological.org
- Faria, N.R., et al. (2021) Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: preliminary findings. virological.org
- Hadfield, J., et al. (2018) Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34(23): 4121-4123. doi:10.1093/bioinformatics/bty407
- Kemp, S.A., et al. (2020a) Neutralising antibodies drive Spike mediated SARS-CoV-2 evasion. medRxiv [Preprint]. doi:10.1101/2020.12.05.20241927
- Kemp, S.A., et al. (2020b) Recurrent emergence and transmission of a SARS-CoV-2 Spike deletion ΔH69/V70. bioRxiv [Preprint]. doi:10.1101/2020.12.14.422555
- Kosakovsky Pond, S.L., et al. (2020) A preliminary selection analysis of the South African V501.V2 SARS-CoV-2 clade. virological.org
- Kovacova, V., et al. (2021) A novel, room temperature-stable, multiplexed RT-qPCR assay to distinguish lineage B.1.1.7 from the remaining SARS-CoV-2 lineages. virological.org
- Larsen, B.B. and Worobey, M. (2020) Identification of a novel SARS-CoV-2 Spike 69-70 deletion lineage circulating in the United States. virological.org
- Larsen, H.D., et al. (2021) Preliminary report of an outbreak of SARS-CoV-2 in mink and mink farmers associated with community spread, Denmark, June to November 2020. Euro Surveill. 26(5): 2100009. doi: 10.2807/1560-7917.ES.2021.26.5.210009
- Lassaunière, R., et al. (2020) SARS-CoV-2 spike mutations arising in Danish mink and their spread to humans. Statens Serum Institut (SSI), Denmark
- McCarthy, K.R., et al. (2020) Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. bioRxiv [Preprint]. doi: 10.1101/2020.11.19.389916
- Moreno, G., et al. (2021) Detection of non-B.1.1.7 Spike ∆69/70 sequences (B.1.375) in the United States. virological.org
- National Institute of Infectious Diseases, NIoID, JAPAN (2021) Brief report: New Variant Strain of SARS-CoV-2 Identified in Travelers from Brazil.
- Naveca, F., et al. (2021a) Phylogenetic relationship of SARS-CoV-2 sequences from Amazonas with emerging Brazilian variants harboring mutations E484K and N501Y in the Spike protein. virological.org
- Naveca, F., et al. (2021b) SARS-CoV-2 reinfection by the new Variant of Concern (VOC) P.1 in Amazonas, Brazil. virological.org
- Oude Munnink, B.B., et al. (2020) Rapid SARS-CoV-2 whole-genome sequencing and analysis for informed public health decision-making in the Netherlands. Nat. Med. 26(9): 1405-1410. doi: 10.1038/s41591-020-0997-y
- Public Health Authority of the Slovak Republic, PHAoSR (2021) Britská mutácia bola na Slovensku už v novembri. V Trenčíne mohla pred sviatkami dominovať (in Slovak).
- Rambaut, A., et al. (2020a) Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. virological.org
- Rambaut, A., et al. (2020b) A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. Nat Microbiol 5: 1403-1407. doi:10.1038/s41564-020-0770-5
- Ren, Y., et al. (2020) The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol Immunol 17: 881-883. doi:10.1038/s41423-020-0485-9
- Resende, P.C., et al. (2020) SARS-CoV-2 genomes recovered by long amplicon tiling multiplex approach using nanopore sequencing and applicable to other sequencing platforms. bioRxiv [Preprint]. doi:10.1101/2020.04.30.069039
- Tegally, H., et al. (2020) Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv [Preprint]. doi:10.1101/2020.12.21.20248640
- Thomson, E.C., et al. (2021) Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell (in press). doi: 10.1016/j.cell.2021.01.037
- Volz, E., et al. (2020) Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. medRxiv [Preprint]. doi: 10.1101/2020.12.30.20249034
- Wang, R., et al (2020) Characterizing SARS-CoV-2 mutations in the United States. Research Square [Preprint]. doi:10.21203/rs.3.rs-49671/v1
- Washington, N.L., et al. (2020) S gene dropout patterns in SARS-CoV-2 tests suggest spread of the H69del/V70del mutation in the US. medRxiv [Preprint]. doi:10.1101/2020.12.24.20248814
- Zhang, C., et al. (2020) Structural basis for the multimerization of nonstructural protein nsp9 from SARS-CoV-2. Mol Biomed 1: 5. doi:10.1186/s43556-020-00005-0