
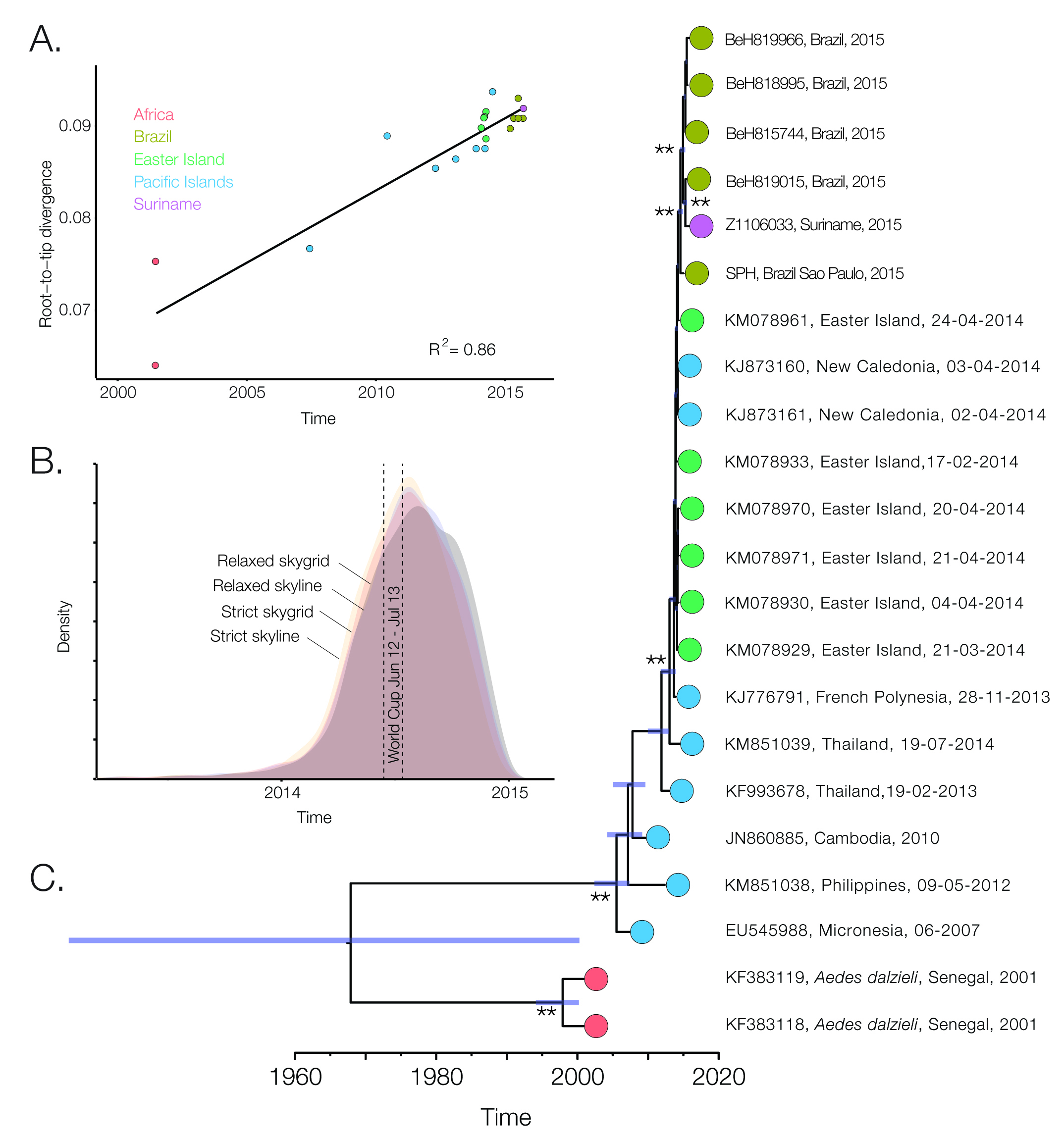

Nice analysis. However;
(1) Please check those Senegal sequences. There are some large outliers in the env and NS5 phylogenies from Senegal which make me strongly suspicious about sequence integrity. I can’t quite tell if they are the ones you have used but please double-check.
(2) Would people have travelled to the World Cup from countries where Zika was previously? Certainly possible, but I would keep an open mind.
The two Senegalese viruses you use are not associated with anomalously long branches are but are reported as recombinant strains in the original Faye et al. (2014) publication. I have excluded these from my trees but I did a quick analysis and KF383118 does appear as recombinant (nature or lab??). Anyway, you might want to use some other Senegalese viruses instead, but I would check them carefully first.
Fantastic work Nuno! And thanks for sharing. However, I just noticed that these sequences are lacking full date information in Genbank. KU365777 is listed with collection_date as 2015. Could you update the Genbank entries and/or list dates for these 4 genomes here?
Question - When running analysis is there a reason that I’m missing for why TMRCA’s would be ‘pulled back in time’ when you run the full Asian/African dataset versus just the Asian dataset in phylogeographic analysis?
I’ve run E gene and full genome for the full dataset (African+Asian) that does not include any of Faye’s sequences and all recombinants as detected by RDPv4 or incongruency also removed. This left 7 African lineage isolates in the analysis and there were 23 isolates in the Asian lineage. Both full genome and E gene analysis agree. TMRCA estimated for Brazil was 2010.5406 [2007.1657, 2013.0183].
However when I ran the Asian lineage alone, again full genome and E gene analysis agree - I got a Brazil TMRCA of 2013.1389 [2012.5698, 2013.8562].
I’m actually interested in TMRCA of our isolates from Thailand, but I noticed that when you run Asian separately you get quite different TMRCAs than when you run African and Asian sequences together. Running the dataset with African isolates pulls the TMRCAs back in time with larger HPDs.
So which analysis would be the more accurate one? When estimating TMRCA is it more important to have more samples and sampling dates or to be lineage specific?
I was unable to get a clock rate for the African lineage alone as I only had 7 samples and didn’t think this would yield an accurate rate. But for the Asian lineage I was getting 8E-4 and when I ran the whole dataset including the African isolates I still got around 8E-4 for meanRate. So I don’t know that I have reason to believe the lineages have/had different rates of evolution and that it would affect TMRCA estimates.
The analysis was lognormal relaxed clock, gamma and CTMC priors were both run and had the same results on clock rates. Model of nt substitution was GTRG. Analysis returned good ESS values from runs of 100 and 250 million iterations. There was nothing in the outputs of any runs that led me to believe one run inferior to the other(s).
Thoughts?