Bas Oude Munnink*, My V.T. Phan*, Sarah C. Hill**, Nuno Rodrigues Faria**, Suzan D. Pas*, Corine H. GeurtsvanKessel*, Chantal B. Reusken*, Marjolein Knoester***, Marjan Wouthuyzen-Bakker***, Wouter F.W. Bierman***, Oliver G. Pybus**, Matthew Cotten*, Marion Koopmans*
*Erasmus MC, Department of Viroscience, Rotterdam, The Netherlands
**Department of Zoology, University of Oxford, United Kingdom
***University of Groningen, University Medical Center Groningen, Department of Medical Microbiology and Infection Prevention, Groningen, The Netherlands
Recently, a new case of yellow fever was diagnosed in a traveller returning from Suriname (7). As this was the first case notified from Suriname in decades, and given the ongoing outbreak of yellow fever in Brazil, we determined the viral genome sequence to seek a possible origin for the virus and to ask if the reported yellow fever virus (YFV) was introduced from the ongoing 2016-2017 Brazilian outbreak (2, 6, 8).
For sequencing, RNA extracted from two serum samples from 04 and 05 March 2017 (both with Ct values of 31), was subjected to reverse transcription and PCR amplified using a set of YFV specific primers spanning the full genome (primer sequences available here: https://github.com/mlcotten/Yellow_fever_virus). PCR products were pooled, sheared, converted to Ion Torrent libraries and sequenced on an Ion Torrent S5XL 530 chip. Short reads were trimmed to remove primers, and low quality positions, de novo assembled into contigs and YFV contigs were identified using standard methods. The genomes from the first round of sequencing had gaps of 30 or 40% of the expected genome length. A second round of sequencing closed one gap and yielded a genome lacking 1600 nucleotides (position 3392-4194 and 6626-7456). The final sequence was submitted to the YFV Typing Tool (1, 3) and genotype assignment was determined to be South America I (SA1), supported with phylogenetic analysis and bootstrap 100.0 (>= 70.0).
The reported genome was aligned with 113 unique YFV sequences from GenBank and with 48 YFV genome sequences from the SA1 genotype, including the most recent YFV sequences from human, primates and mosquitoes in Brazil (8). Molecular phylogenies of global YFV and of the SA1-specific lineage were inferred in MrBayes v.3.2.3 running with three independent Bayesian Markov Chain Monte Carlo (BMCMC) chains, chain length of 1 million generations, employing the GTR+Γ4 model of substitution. The resulting trees were visualized in FigTree (5), mid-point rooted for clarity and posterior probabilities >= 0.75 were shown (Figure 1 and 2).
The reported YFV sequence fits into the SA1 clade in the global phylogeny of YFV, consistent with the Yellow Fever Virus genotyping results (Figure 1). Within the SA1 lineage (Figure 2), the Surinamese YFV sequence was most closely related to Brazilian sequences identified in 2000 and 2001 from human and mosquito hosts (4) (Figure 2). The current Brazilian YFV outbreak sequences belonged to a different lineage that includes sequences identified in human, primates and mosquitoes (Figure 2). This suggests that the YFV case from Suriname is not directly related to the current (2016-2017) YFV outbreak in the southeast of Brazil. This also shows the importance of detailed genomic surveillance, as background for outbreak investigations. Continued presence of YFV in an enzootic cycle leads to recognizable geographical signatures that can be used to trace origins of outbreaks. However, systematic surveillance and sharing of YFV genomic data is needed from other regions in the Americas including the Amazon to better define these patterns.
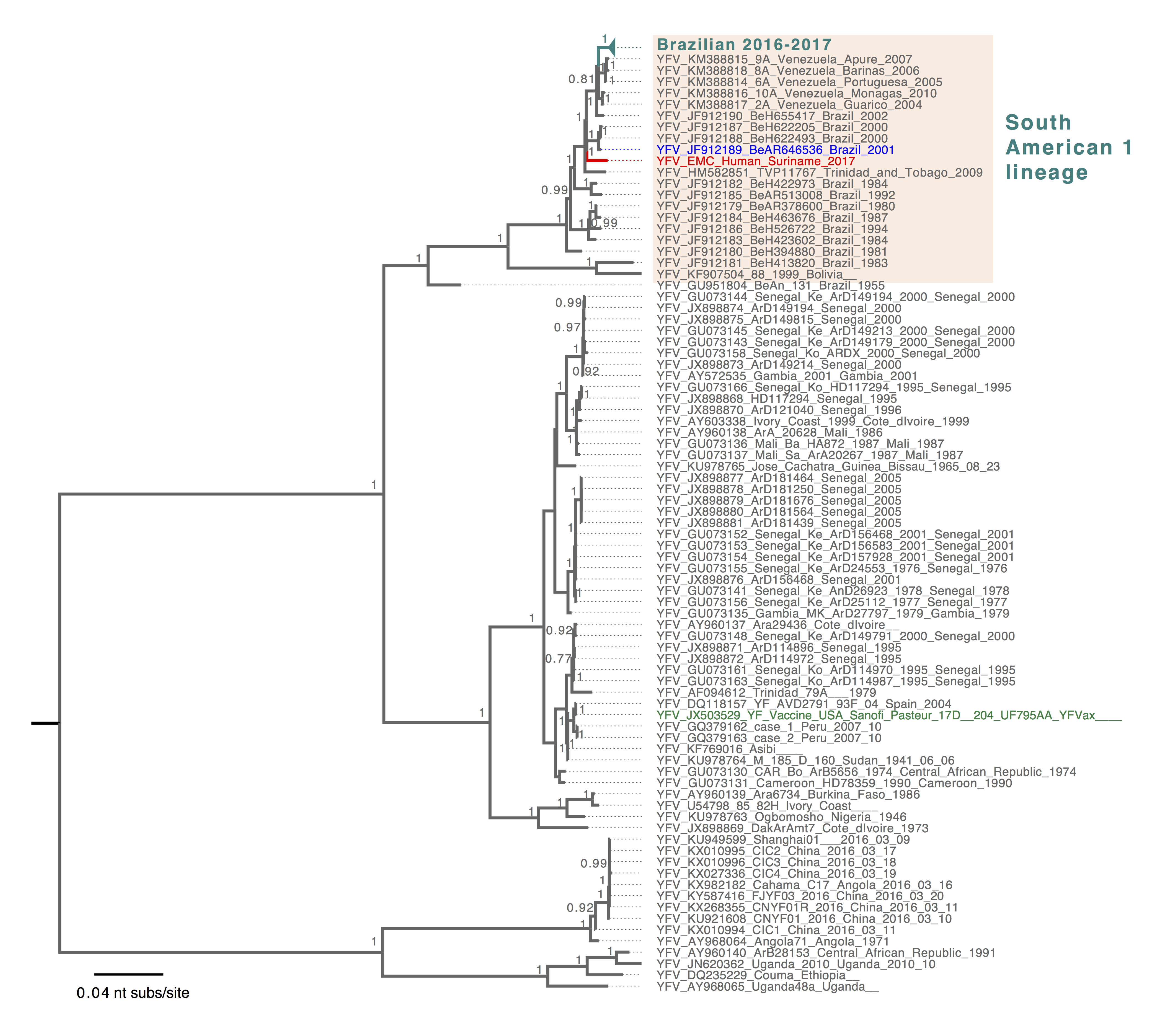
Figure 1. Bayesian phylogenetic tree of global YFV sequences. The South American 1 genotype sequences were shaded in light orange. The reported Surinamese YFV was highlighted in red, and YFV sequence identified from mosquito was highlighted in blue and the YFV vaccine strain sequence was highlighted in green. Tree was mid-pointed rooted and only posterior probabilities >= 0.75 were shown. The scale bar was drawn to number of nucleotide substitutions per site.

Figure 2. Bayesian phylogeny of the YFV South American 1 lineage. The lineage containing YFV sequences from the ongoing outbreak in Brazil was collapsed and highlighted in turquoise, and Brazilian YFV sequences from earlier years that are most closely related to the reported Surinamese YFV were shaded in light pink. The reported Surinamese YFV was highlighted in red, and YFV sequences identified from mosquito was highlighted in blue. Tree was mid-pointed rooted and only posterior probabilities >= 0.75 were shown. The scale bar was drawn to number of nucleotide substitutions per site.
Acknowledgements The sequencing work was supported by the European Union’s Horizon 2020 research and innovation programme under grant agreements No 643476 (COMPARE) and No 634650 (Virogenesis) and is part of the WHO Collaborating Centre for Arbovirus and Haemorrhagic Fever Reference and Research.
Note We are currently in the process of preparing GenBank and Short Read Archive submissions. If you would like to use this sequence please contact us directly.
References
- Alcantara, L., N. Faria, P. Libin, M. Nunes, V. Fonseca, M. I. Restovic, M. Freire, M. Giovanetti, K. Theys, L. Cuypers, A. Nowé, E. Vanden Eynden, A. Abecasis, K. Deforche, G. Santiago, I. de Siqueira, J. Vasconcelos, R. da Cunha, O. Pybus, A.-M. Vandamme, and T. de Oliveira. 2017. An automated method for the identification of Dengue, Zika, Yellow Fever and Chikungunya virus species and genotypes. https://lirias.kuleuven.be/handle/123456789/574421.
- Bonaldo, M. C., M. M. n. Gómez, A. AC dos Santos, F. V. Santos de Abreu, A. Ferreira-de-Brito, R. Moraes de Miranda, M. G. a. de Castro, and R. Lourenço-de-Oliveira. 2017. Genome analysis of yellow fever virus of the ongoing outbreak in Brazil reveals polymorphisms. Mem Inst Oswaldo Cruz, Rio de Janeiro 112:1-5. http://memorias.ioc.fiocruz.br/issues/past-issues/item/6275-000134_genome-analysis-of-yellow-fever-virus-of-brazil-ongoing-outbreak-reveals-polymorphisms
- Fonseca, V., L. C. J. Alcontara, and T. de Oliveira. 2016. Yellow Fever Virus Typing Tool (2016). www.bioafrica.net/software.php.
- Nunes, M. R., G. Palacios, J. F. Cardoso, L. C. Martins, E. C. Sousa, Jr., C. P. de Lima, D. B. Medeiros, N. Savji, A. Desai, S. G. Rodrigues, V. L. Carvalho, W. I. Lipkin, and P. F. Vasconcelos. 2012. Genomic and Phylogenetic Characterization of Brazilian Yellow Fever Virus Strains. Journal of Virology, 86(24), 13263–13271. http://doi.org/10.1128/JVI.00565-12
- Rambaut, A. 2016. FigTree-v1.4.3. http://tree.bio.ed.ac.uk/software/figtree/.
- WHO. 2017. Yellow fever – Brazil Disease outbreak news 4 April 2017. http://www.who.int/csr/don/04-april-2017-yellow-fever-brazil/en/.
- Wouthuyzen-Bakker M., Knoester M., van den Berg A.P., GeurtsvanKessel C.H., Koopmans M.P., Van Leer-Buter C., Oude Velthuis B., Pas S.D., Ruijs W.L., Schmidt-Chanasit J., Vreden S.G., van der Werf T.S., Reusken C.B., Bierman W.F. Yellow fever in a traveller returning from Suriname to the Netherlands, March 2017. Euro Surveill. 2017;22(11):pii=30488. DOI: http://dx.doi.org/10.2807/1560-7917.ES.2017.22.11.30488
- YiBRA_consortium. 2017. Real-time Genomic Surveillance of the Yellow Fever Virus Outbreak in Brazil, 2017 http://virological.org/t/real-time-genomic-surveillance-of-the-yellow-fever-virus-outbreak-in-brazil-2017/464.