Matteo Chiara1, Pietro Pinoli2, Luca Minotti2, Anna Bernasconi2, Arif Canakoglu3, Erica Ferrandi4, and Stefano Ceri2
1 Dipartimento di Bioscienze, Università degli Studi di Milano, 20133 Milan, Italy
2 Dipartimento di Elettronica, Informazione e Bioingegneria, Politecnico di Milano, 20133, Milan, Italy
3 Dipartimento di Anestesia, Rianimazione ed Emergenza-Urgenza, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy
4 Istituto di Biomembrane e Bioenergetica, CNR, 70126 Bari, Italy
VariantHunter (http://gmql.eu/variant_hunter) is a novel tool to analyze the frequencies of amino acid changes in SARS-CoV-2 at the region/country/continent levels to capture interesting trends in viral genome evolution and/or identify novel emerging variants. VariantHunter incorporates data from GenBank, curated and made openly available by Nextstrain (at https://nextstrain.org/ncov/open/global) and regularly updated. A Docker implementation of VariantHunter that allows users to analyze restricted-access data (i.e., from GISAID) or private data is also available.
VariantHunter supports two main modes of analysis: lineage dependent and lineage independent. Both methods analyze a time interval of four weeks and perform a simple incremental analysis of the prevalence of amino acid changes in time, to pinpoint allele frequencies in the viral population, which might subtend the emergence of a novel variant. Previous experience with SARS-CoV-2 has clearly demonstrated how the emergence and spread of novel variants are associated with a rapid surge in the prevalence of a series of amino acid changes in the viral population.
VariantHunter lineage independent mode, which performs global analyses of allele frequency at a user-specified geographic location, can clearly capture these patterns, as illustrated by a selection of use cases presented below. The lineage dependent mode, which performs equivalent analyses but only in a specific lineage, has instead been designed to facilitate the tracking/identification of emerging sub-lineages of the virus with a potential relative growth advantage with respect to their parental lineage.
For a complete explanation of the rationale behind the tool and of its main outputs, readers may refer to the documentation available here. In the following, we report a selection of 6 use cases to illustrate VariantHunter’s potential. Cases 1-5 were extracted using GenBank data [1], Case 6 using GISAID data [2].
Case 1. Emergence and spread of the Alpha variant in the UK in October/November 2022.
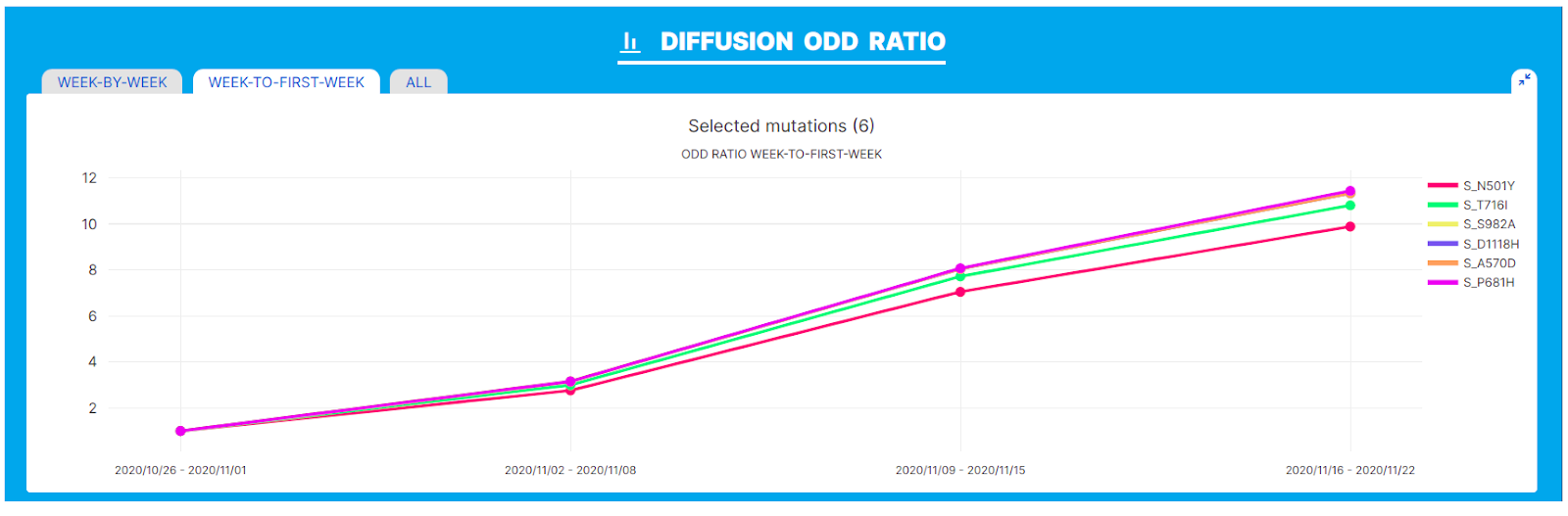
VariantHunter lineage independent analysis was applied to an interval of 4 weeks spanning from 26/10/2020 to 16/11/2020 in the UK. This interval corresponds with the emergence and spread of the Alpha variant. The main outputs of VariantHunter clearly capture the trends associated with the emergence of a novel variant. For example, in the ‘Diffusion Odd Ratio’ chart below, 6 amino acid changes in the Spike glycoprotein are singled out due to their remarkable increase in frequency (from 1% to 11%). Importantly, the 6 amino acid changes highlighted in the plot account for the complete set of amino acid changes in Spike of the Alpha SARS-CoV-2 variant.
Case 2. Early spread of the Delta variant in the UK in April/May 2021.
We analyzed an interval of 4 weeks spanning from 10/04/2021 to 07/05/2021 in the UK, corresponding with the early spread of the Delta variant in the UK.
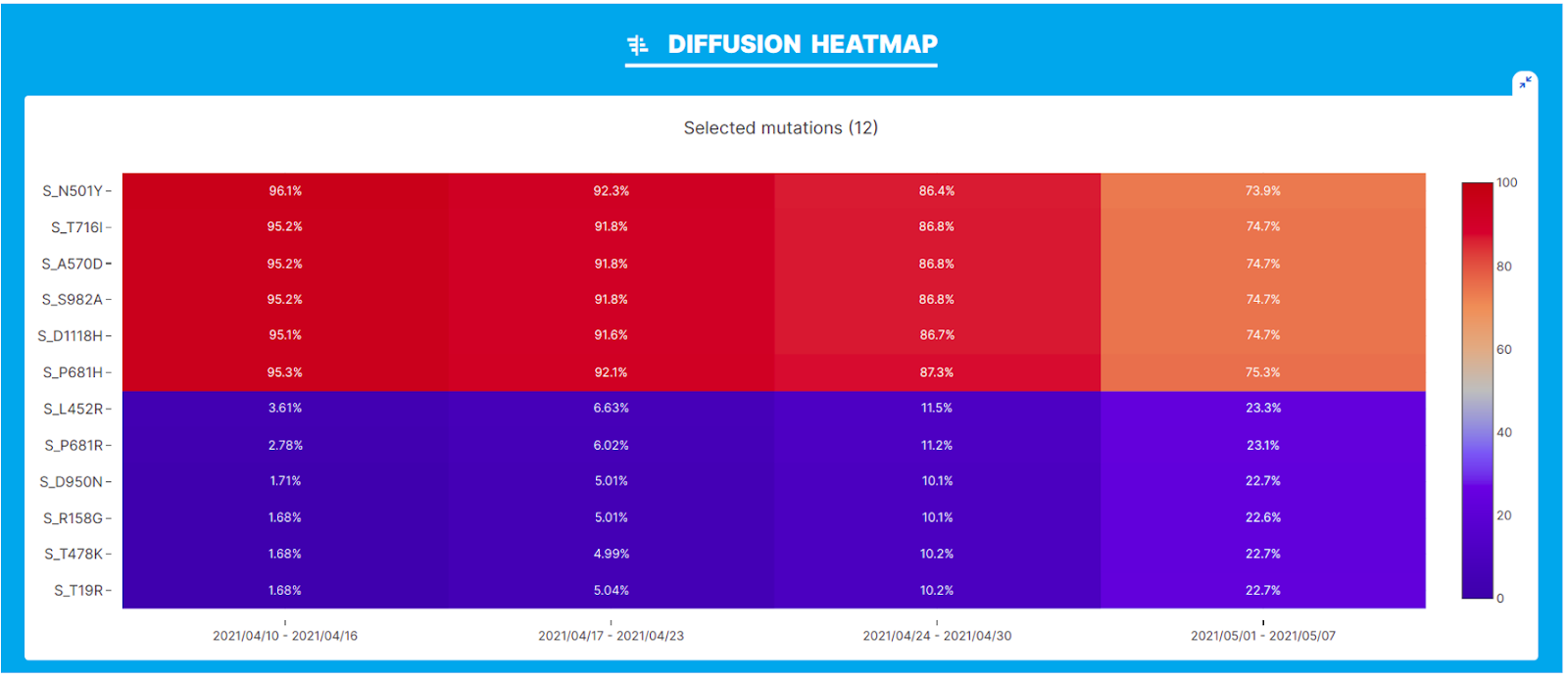
VariantHunter’s ‘Diffusion Heatmap’ illustrates the prevalence of 6 amino acid changes in the Spike glycoprotein characteristic of the Alpha variant (N501Y, T716I, A570D, S982A, D1118H, P681H) and a matched number of amino acid changes characteristic of the Delta variant (L452R, P681R, D950N, R158G, S478K, T19R). A clear increase in the changes associated with the Delta variant and a concomitant decrease in the changes characteristic of the Alpha variant can be observed.
As a useful complement to the analysis, the mutation table reported below illustrates the relative prevalence/association of S:N501Y (mostly associated with Alpha-related lineages) and S:L452R (observed almost exclusively in lineages/sub-lineages of Delta). From this table it is possible to appreciate how in the early phase of spread of the Delta variant, L452R was mainly associated with the B.1.617.1 and B.1.617.2 lineages.

Case 3. Spread of the Omicron variant and concomitant displacement of the Delta variant.
To further illustrate the functionalities of VariantHunter we ran the analysis in Europe and North America, considering the interval of time spanning from 05/12/2021 to 01/01/2022, which coincides with the rapid emergence and spread of the Omicron variant of SARS-CoV-2 worldwide.
A clear decrease in the prevalence of 6 amino acid changes characteristic of the Delta variant (L452R, P681R, D950N, R158G, S478K, T19R) and a concomitant and striking increase of 23 amino acid changes associated with the Omicron variant of SARS-CoV-2 is observed both in Europe and North America. The greater uniformity of plots describing mutation trends in Europe (left) vs North America (right) may indicate greater accuracy in sequencing processes.

Using the lineage specific analysis, VariantHunter can also be used to detect the rapid increase in prevalence of a variant of SARS-CoV-2 in a pre-existing lineage, and hence facilitate the identification/detection of novel sub-lineages with a potential growth advantage. To illustrate the application of VariantHunter in this scenario, we apply our tool to replicate the main findings of 3 Pango designation issues [3] that led to the definition of novel sub-lineages of SARS-CoV-2. Specifically, we refer to the issues 499, 508, and 394.
Case 4. The Omicron BA.2.12.2 lineage.
Pango designation issue 499 led to the definition of the BA.2.12.2 sub-lineage of SARS-CoV-2. The novel lineage is defined by a L452Q amino acid change in the spike glycoprotein according to the submitter.
We performed our analysis on the BA.2.12 lineage in the geographic location USA and interval of time March 2022, where the novel variant was first described. A clear increase in the prevalence of L452Q within lineage BA.2.12.2 is observed both from the ‘Diffusion Heatmap’ and the 'Diffusion Odds Ratio’ chart. As outlined in the latter, the amino acid change that defines the new lineage seems to be associated with a clear growth advantage.
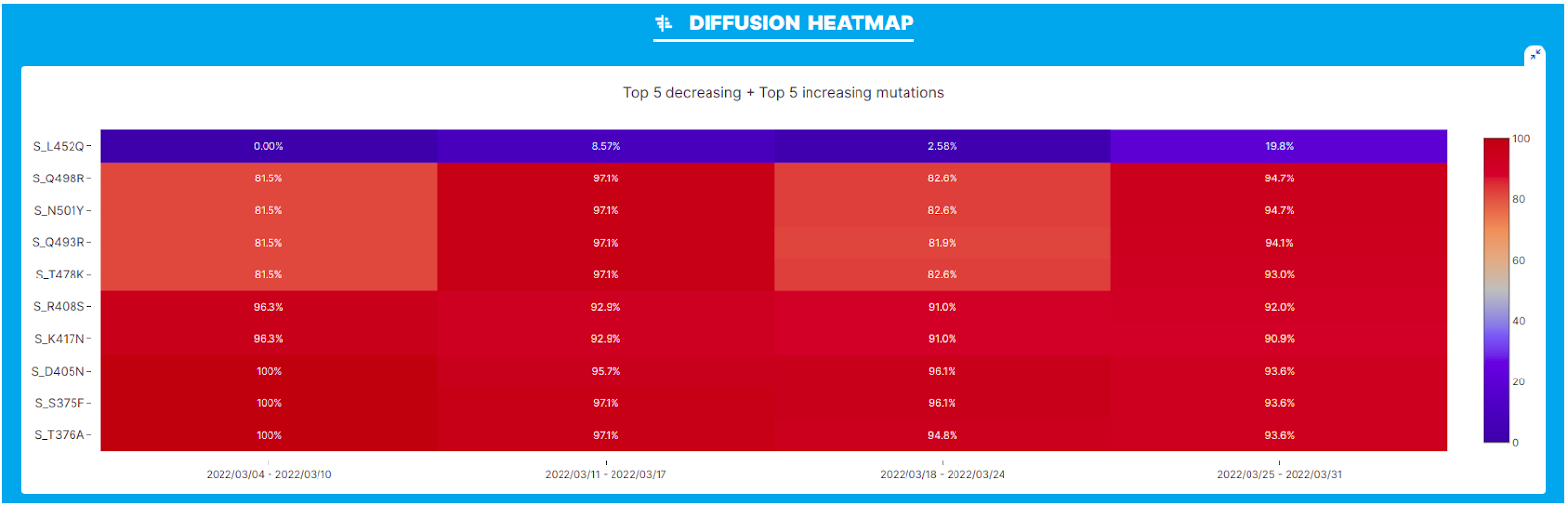
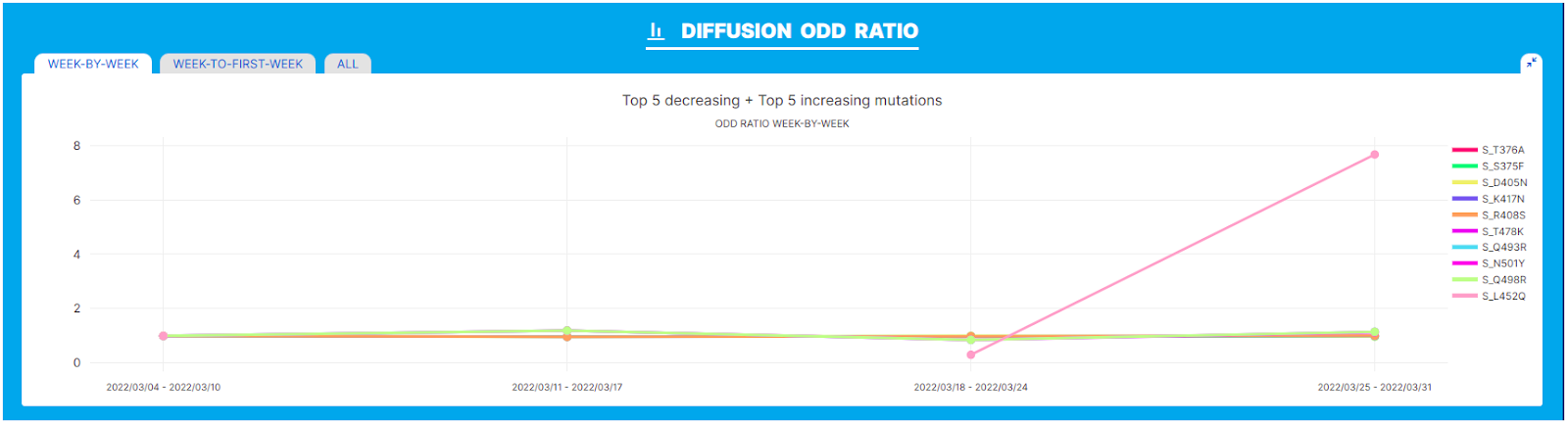
[Note that in this plot values around 1 indicate a steady state: no increase or decrease in frequency, values below 1 indicate a decrease, and values > 1 are indicative of rapid increase/relative growth advantage)]
Case 5. The Omicron BA.1.15.2 lineage.
Similarly, VariantHunter can also confirm the findings reported by the Pango designation issue 508, which led to the definition of the BA.1.15.2 sub-lineage of SARS-CoV-2. In this case, the novel lineage is defined by a Q628K amino acid change in the spike glycoprotein according to the submitter.
We performed our analysis on the BA.1.15 lineage in the geographic location USA and interval of time March 2022, corresponding with the emergence of BA.1.15.2. Again, a clear increase in the prevalence of Q628K within lineage BA.1.15.2 is observed by the plots below.
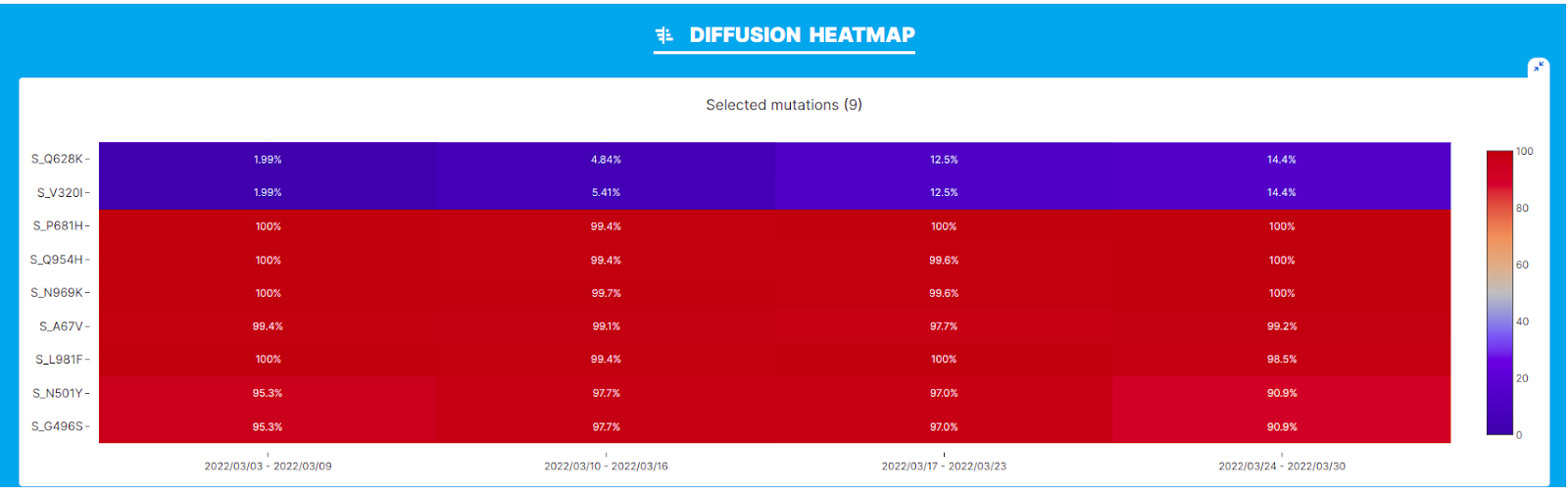

Interestingly, we also observe a similar increase in the prevalence of S:V320I, an amino acid change that to the best of our knowledge is not currently associated with/does not define any sub-lineage of BA.1.15. Both S:V320I and S:Q628K show a similar relative growth advantage according to the ‘Diffusion Odds Ratio’ chart compared to the amino acid changes that define the parental lineage. After a rapid surge in frequency from 1 to 12%, the two novel mutations seem to ‘stabilize’ around 15%, in the last week of the interval considered in the analysis, and show an odd ratio of ~1.
Case 6. The Delta AY.122.6 lineage.
Finally, we also show how VariantHunter can reproduce the findings reported by the Pango designation issue 394, which lead to the definition of the AY.122.6 sub-lineage of the Delta SARS-CoV-2 variant. The novel lineage is defined by the E484A amino acid change in the Spike glycoprotein according to the submitter and shows a relative growth advantage in France compared to the parental lineage AY.122.
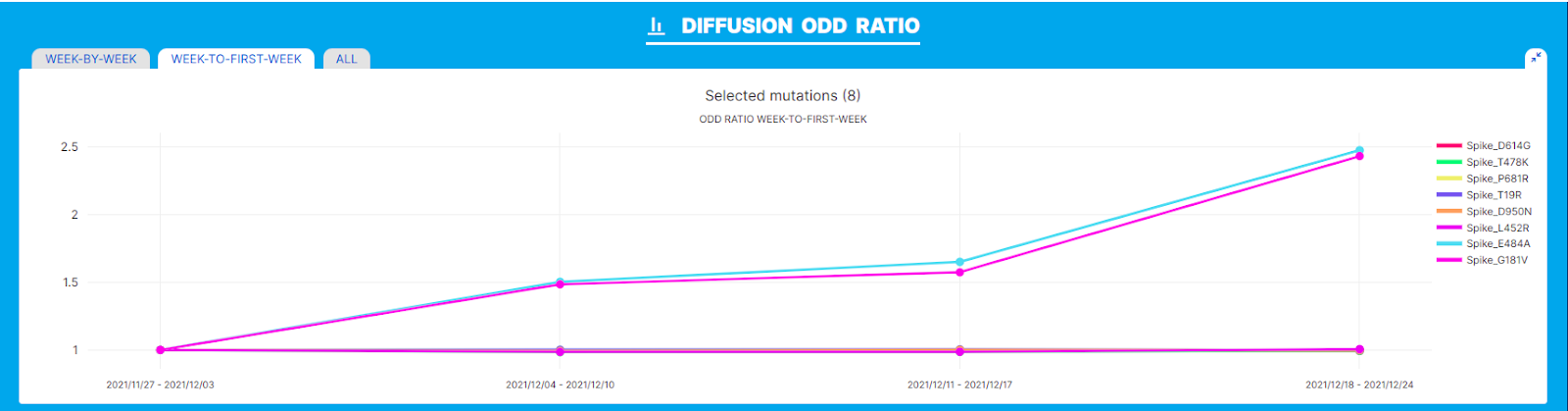
We performed our analysis on the AY.122 lineage in the geographic location France and interval of time November/December 2021, associated with the emergence of AY.122.6. A clear increase in the prevalence of S:E484A and of another amino acid change in Spike, S:181V, can be observed within lineage AY.122. According to the diffusion heatmap both changes show a consistent increase in prevalence (from 10% to 26%) in the interval of time included in the analysis.

The 'Diffusion Odds Ratio’ chart is consistent with a relative advantage in growth for both amino acid changes (S:E484A and S:G181V), as illustrated by the constant increase of the ‘diffusion’ odd ratio in the corresponding plot and by the fact that odd ratio values > 1 are recovered at every time point of the 4 weeks interval considered in the analysis. Indeed, in this plot values around 1 indicate a steady state (no increase or decrease in prevalence), values below 1 indicate a decrease, and values > 1 indicate a rapid increase/relative growth advantage.
Notes for users
VariantHunter Docker version is optimized for being used with big datasets (e.g., ~10M sequences on GISAID as of May 2022, which can be downloaded as a metadata.tsv file from users with an enabled account). Complete documentation of the described cases can be found at http://gmql.eu/variant_hunter.
References
[1] Eric W Sayers et al. Genbank. Nucleic acids research, 49(D1):D92–D96, 2021.
[2] Yuelong Shu and John McCauley. GISAID: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance, 22(13), 2017.
[3] Áine O’Toole et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evolution, 7(2):veab064, 2021.