MPXV_schema.new_nom.pdf (311.0 KB)
Urgent need for a non-discriminatory and non-stigmatizing nomenclature for monkeypox virus
Authors: Christian Happi1,2*, Ifedayo Adetifa3, Placide Mbala4, Richard Njouom5, Emmanuel Nakoune6, Anise Happi1, Nnaemeka Ndodo3, Oyeronke Ayansola3, Gerald Mboowa7, Trevor Bedford8,9, Richard A. Neher10,11, Cornelius Roemer10,11, Emma Hodcroft11,12,13, Houriiyah Tegally14,15, Áine O’Toole16, Andrew Rambaut16, Oliver Pybus17,18,19, Moritz U.G. Kraemer17,18, Eduan Wilkinson14, Joana Isidro20, Vítor Borges20, Miguel Pinto20, João Paulo Gomes20, Cheryl Baxter15,21, Richard Lessells14,21, Ahmed E. Ogwell7, Yenew Kebede7, Sofonias K. Tessema7, Tulio de Oliveira14,15,21,22*
Affiliations:
1 African Centre of Excellence for Genomics of Infectious Diseases (ACEGID), Redeemer’s University; Ede, Osun State, Nigeria
2 Department of Biological Sciences, Faculty of Natural Sciences, Redeemer’s University, Ede, Osun State, Nigeria.
3 Nigeria Centre for Disease Control, Abuja, Nigeria
4 Institut National de Recherche Biomedicale, Kinshasa, Democratic Republic of the Congo; University of Kinshasa, Democratic Republic of Congo;
5 Virology Unit, Centre Pasteur of Cameroon;
6 Institut Pasteur Bangui, Central African Republic
7 Africa Centres for Disease Control and Prevention (Africa CDC), Addis Ababa, Ethiopia
8 Vaccine and Infectious Disease Division, Fred Hutchinson Cancer Center, Seattle, WA, USA
9 Howard Hughes Medical Institute, Seattle, WA, USA
10 Biozentrum, University of Basel, Switzerland
11 Swiss Institute of Bioinformatics, Lausanne, Switzerland
12 Institute of Social and Preventive Medicine, University of Bern, Bern, Switzerland
13 HUG Virology Laboratory, University of Geneva, Geneva, Switzerland
14 KwaZulu-Natal Research Innovation and Sequencing Platform (KRISP), Nelson R. Mandela School of Medicine, University of KwaZulu-Natal, Durban, South Africa;
15 Centre for Epidemic Response and Innovation (CERI), School of Data Science and Computational Thinking, Stellenbosch University, Stellenbosch, South Africa;
16 Institute of Ecology and Evolution, University of Edinburgh, Edinburgh, UK;
17 Department of Zoology, University of Oxford, Oxford, UK;
18 Pandemic Sciences Institute, University of Oxford, UK;
19 Department of Pathobiology and Population Sciences, Royal Veterinary College, London, UK
20 Genomics and Bioinformatics Unit, Department of Infectious Diseases, National Institute of Health (INSA), Lisbon, Portugal
21 Centre for the AIDS Programme of Research in South Africa (CAPRISA), Durban, South Africa;
22 Department of Global Health, University of Washington, Seattle, WA, USA*Correspondence: Christian Happi ([email protected]), Tulio de Oliveira ([email protected])
Background
Monkeypox is a disease caused by the monkeypox virus (MPXV) from the Orthopoxvirus genus in the family Poxviridae [1,2]. Since the first report of monkeypox virus infection in humans in the 1970s [3], repeated outbreaks have been reported periodically in Western and Central Africa and global events have been detected rarely [4,5]. However, a recent global outbreak of MPVX has been detected without a clear link to Africa [6]. As of 8 June 2022, at least 1111 human cases of MPXV have been confirmed or suspected and cases have been detected in 44 countries [7]. MPXV infection is caused normally by spill-over events to humans from animals such as rodents, squirrels, and non-human primates [1,4,5]. The virus can be also transmitted from one person to another by close contact with lesions, body fluids, respiratory droplets and contaminated materials [1,4]. Case counts and epidemiological patterns suggest that the current global outbreak is sustained by human-to-human transmission [6,8].
The prevailing perception in the international media and scientific literature is that MPXV is endemic in people in some African countries. However, it is well established that nearly all MPXV outbreaks in Africa prior to the 2022 outbreak, have been the result of spillover from animals to humans and only rarely have there been reports of sustained human-to-human transmissions. In the context of the current global outbreak, continued reference to, and nomenclature of this virus being African is not only inaccurate but is also discriminatory and stigmatizing. The most obvious manifestation of this is the use of photos of African patients to depict the pox lesions in mainstream media in the global north. Recently, Foreign Press Association, Africa issued a statement urging the global media to stop using images of African people to highlight the outbreak in Europe [9].
Although the origin of the new global MPXV outbreak is still unknown, there is growing evidence that the most likely scenario is that cross-continent, cryptic human transmission has been ongoing for longer than previously thought. However, there is an increasing narrative in the media and among many scientists that are trying to link the present global outbreak to Africa or West Africa, or Nigeria. Further, the use of geographical labels for strains of MPXV, specifically, references to the 2022 outbreak as belonging to the “West African” or “Western African” clade, strain, or genotype. We therefore believe that a nomenclature that is neutral, non-discriminatory and non-stigmitizing will be more appropriate for the global health community.
Current classification
In the current classification of MPXV genetic diversity only two clades of MPXV are recognized – referred to as the “West African” clade and the “Central African” or “Congo Basin” clade[10]. However, these historic MPXV clade names are counter to the best practice of avoiding geographic locations in the nomenclature of diseases and disease groups [11,12]. The recent and prompt example implemented for SARS-CoV-2 should be the norm [11]. Given the increasingly rapid communication of, and attention to, the international human MPXV outbreak, it is important to consider an appropriate, non-discriminatory, and non-stigmatizing nomenclature and classification of MPXV clades. In recent publications[12] and symposia, including the WHO Research and Development (R&D) symposium, it was highlighted that the current global outbreak was caused by MPXV of the West African clade. Some genome sequences on the NCBI Genbank database use “West African” for the field “strain” or “genotype” (including the NCBI reference genome: NC_063383). Like many previous geographic labels of infectious diseases based on locations of first detection, it is misleading and inaccurate because very limited surveillance and limited diagnostic capacity means that the full range of the pathogen is not known. This is crucially demonstrated by the discovery in May 2022 that MPXV has been circulating in over 44 countries without detection and is likely to be present in many more.
Proposed Classification
Here, we propose a novel classification of MPXV that is non-discriminatory and non-stigmatizing and aligned with best practices in naming of infectious diseases [11] in a way that minimizes unnecessary negative impacts on nations, geographic regions, economies and people and that considers the evolution and spread of the virus.
In the original proposal, we named MPXV clades 1,2,3, in order of detection, but in discussions with expert committees of the WHO, we agreed to name these as clades I, IIa, IIb to represent the historical discovery of the MPVX. These clades include viral genomes from Western African, Central African and localized spillover events in global north countries and from both human and non-human hosts (Figure 1A). Here, clade I corresponds to the prior “Congo Basin clade”, while clades IIa and IIb corresponds to the prior “West African clade”. These three clades represent deep MPXV diversity, accumulated over many years of evolution in the animal reservoir. Further sequencing of MPXV from the animal reservoir may potentially uncover further clades III, IV, V, and so forth.
We also suggest naming a new sub-clade of IIb, containing genomes sampled between 2017–2019 from the UK, Israel, Nigeria, USA, and Singapore and genomes from 2022 global outbreaks (Figure 1B). Since viruses in this clade have been transmitting from person to person in dozens of countries and potentially over multiple years, we propose that this represents transmission route distinct from that of previous MPXV cases in humans and should be afforded a distinct name so that it can be referred to specifically in both scientific discourse and the general media. Whilst the formal naming of virus species is the purview of the International Committee of Taxonomy of Viruses (ICTV), we believe this is an opportunity for a break with the name monkeypox and the historical associations attached to that name. However, we believe that a distinct and convenient name for the virus causing this epidemic would facilitate communication without further negative connotations. Here we use the placeholder label ‘hMPXV1’ to denote where we believe this now human virus becomes distinct from MPXV [13] (Figure 1B), and urge a speedy decision and adoption of a new name.
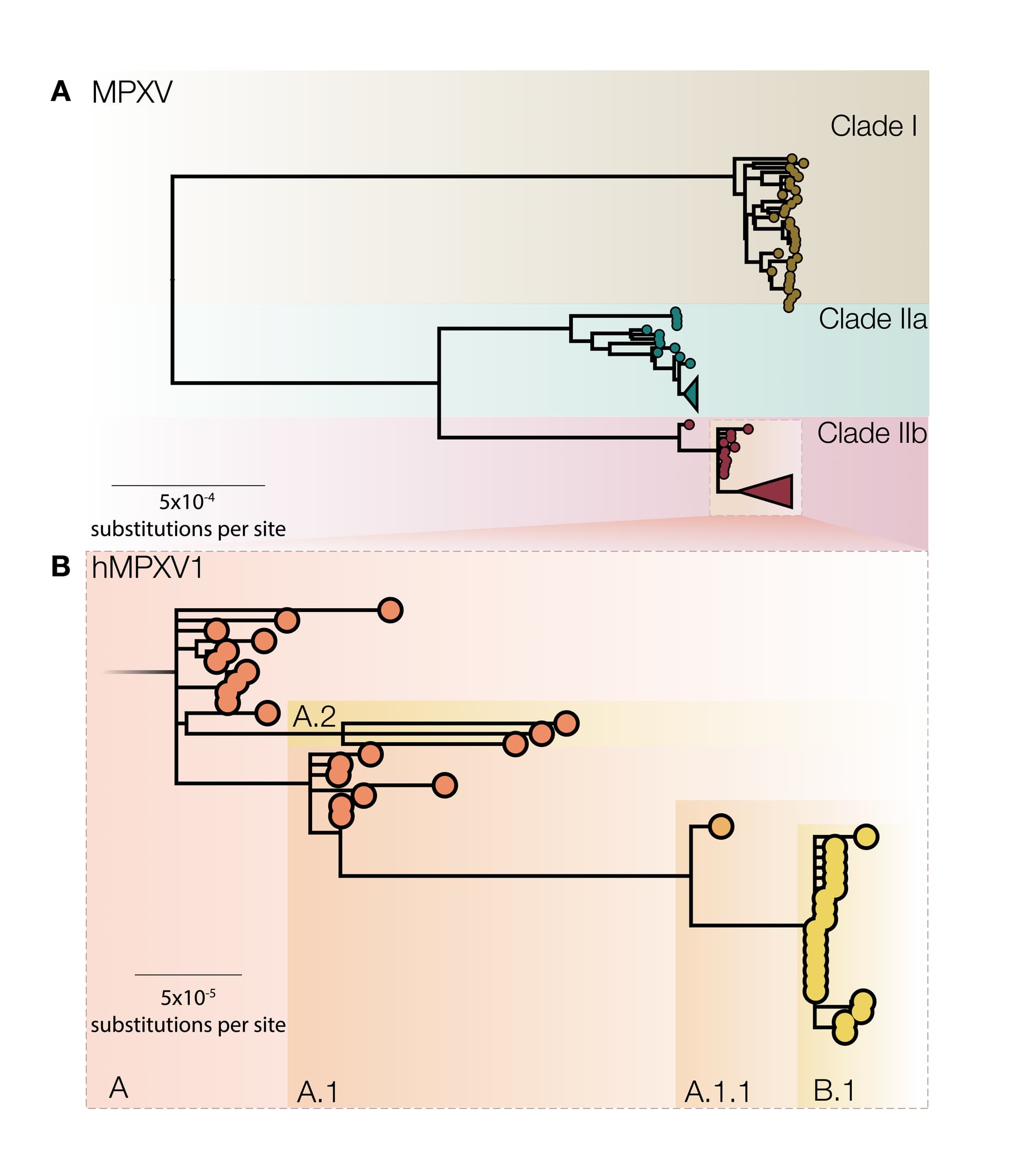
Figure 1 | A) A midpoint rooted maximum likelihood (iqtree2 using JC model) phylogeny of MPXV genomes sampled from human and non-human infections in 1970-2022 aligned against the reference genome (accession NC_063383) with one of the ITR regions (from 190788 onwards in the genome). A number of repetitive regions were also masked out. Three distinct MPXV clades are indicated, representing the deep diversity of MPXV. Clade I corresponds to the prior “Congo Basin clade”, while Clades IIa and IIb corresponds to the prior “West African clade”. Clade 3 contains most genomes from the 2017, 2018 and 2022 human outbreaks. B) Proposed nomenclature for genomes belonging to the 2017 - 2019 outbreaks from the UK, Israel, Nigeria, USA, and Singapore and genomes from 2022 global outbreaks as a broader classification of the ‘hMPXV1’ virus with its diversity denoted by neutral lineages such as A, A.1, A.1.1, B.1, etc.
Within the hMPXV1 sub-clade there is already notable diversity even amongst the limited number of genomes so far described. Thus we further propose that distinct lineages and clades within the epidemic are given neutral names and suggest a system similar to Pango nomenclature for SARS-CoV-2 [14] with lineages within the hMPXV clade given labels that encode genealogical relationships. To keep the labels short, we propose to introduce aliases after the second subdivision, instead of after three as in the original Pango scheme. Under this nomenclature, the base of hMPXV1 would be denoted lineage ‘A’, the descendant lineages would be named as ‘A.1’, ‘A.2’, ‘A.1.1’ and the current international 2022 outbreak would be denoted ‘B.1’ as the first detected descendent lineage of ‘A.1.1’ (Figure 1B). We urge the international community to adopt such a system to preempt the adoption of informal and potentially stigmatizing labels.
With the above suggestions, we encourage the community to adopt a principled and neutral naming scheme such as the one presented here. We believe that this new classification will be easily adopted and is supported by the Africa Centre for Diseases Control and Prevention (Africa CDC) and the World Health Organization (WHO) [15] .
We hope that the world uses the current outbreak to advance our understanding, and provides the funding and focus for effective regional and global public health surveillance for emerging and re-emerging threats. By supporting a non-discriminatory and non-stigmatizing classification, we can encourage African and other researchers in low- and middle-income countries (LMICs) to advance genomic surveillance, share sequence data, and minimize negative impacts. Failure to support and adopt the proposed nomenclature and classification may result in loss of interest in sustaining active surveillance and rapid reporting of pathogens with epidemic and pandemic potentials, by scientists and national public health institutions in Africa and other LMICs. Every case of MPXV infection should be treated with the same attention and sense of urgency as the ones now in European countries and North America. The entire epidemic of hMPXV1 regardless of the location needs to be halted, not just this Northern hemisphere outbreak. A practical and neutral system of nomenclature allows efficient communication without the risk of further misconceptions, discrimination and stigmatisation.
References:
-
World Health Organization. Monkeypox fact sheet. Available from: https://www.who.int/news-room/fact152sheets/detail/monkeypox. Accessed: 3 June 2022. Geneva, Switzerland: WHO, 2019.
-
European Centre for Disease Prevention and Control. Monkeypox multi-country outbreak – 23 May 2022. Available from: Risk assessment: Monkeypox multi-country outbreak. Accessed: 3 June 2022. Stockholm: European Centre for Disease Prevention and Control, 2022.
-
Ladnyj ID, Ziegler P, Kima E. A human infection caused by monkeypox virus in Basankusu Territory, Democratic Republic of the Congo. Bull World Health Organ. 1972;46(5):593-597.
-
Yinka-Ogunleye A, Aruna O, Dalhat M, et al. Outbreak of human monkeypox in Nigeria in 2017-18: a clinical and epidemiological report. Lancet Infect Dis 2019; 19(8): 872-9.
-
Bunge EM, Hoet B, Chen L, et al. The changing epidemiology of human monkeypox-A potential threat? A systematic review. PLoS Negl Trop Dis 2022; 16(2): e0010141.
-
Vivancos R, Anderson C, Blomquist P, et al.Eurosurveillance | Community transmission of monkeypox in the United Kingdom, April to May 2022. Eurosurveillance 2022; 27(22): 2200422.
-
Kraemer MUG, Tegally H, Pigott DM, et al. Tracking the 2022 monkeypox outbreak with epidemiological data in real-time. June 08, 2022. The Lancet Infectious Diseases. DOI:Redirecting
-
Vivancos R, Anderson C, Blomquist P, et al. Community transmission of monkeypox in the United Kingdom, April to May 2022. Eurosurveillance 2022; 27(22): 2200422.
-
Wandera K, Okwach D, Morgan H. Our statement on the use of black people to depict outbreak of monkeypox in Europe and North America. Available from: https://twitter.com/FPA_Africa/status/1527990596044001282. Accesssed: 7 June 2022. Nairobi, Kenya: Foreign Press Association, Africa, 2022.
-
Likos AM, Sammons SA, Olson VA, et al. A tale of two clades: monkeypox viruses. J Gen Virol 2005; 86(Pt 10): 2661-72
-
Konings F, Perkins MD, Kuhn JH, et al. SARS‐CoV‐2 variants of interest and concern naming scheme conducive for global discourse.Nat Microbiol. 2021;6:821‐823.
-
World Health Organization. World Health Organization Best Practices for the Naming of New Human Infectious Diseases. Available from: https://apps.who.int/iris/bitstream/handle/10665/163636/WHO_HSE_FOS_15.1_eng.pdf. Accessed: 6 June 2022. Geneva, Switzerland: World Health Organization, 2015.
-
O’Toole A, Rambaut A. Initial observations about putative APOBEC3 deaminase editing driving short-term evolution of MPXV since 2017. Available from: https://pando.tools/t/initial-observations-about-putative-apobec3-deaminase-editing-driving-short-term-evolution-of-mpxv-since-2017/830. Accessed: 7 June 2022. ARTIC Network, 2022.
-
Rambaut A, Holmes EC, O’Toole Á, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol. 2020;5(11):1403–7
-
World Health Organization. Monkeypox: experts give virus variants new names. Available from: Monkeypox: experts give virus variants new names. Accessed: 16 August 2022. Geneva, Switzerland: World Health Organization, 2022.
Supplementary Material
A NextStrain view of the proposed nomenclature
Supplementary Table 1. MPXV genomes
| Accession | Clade | Lineage | Country | Year | Reference |
|---|---|---|---|---|---|
| DQ011156 | 2 | Liberia | 1970 | (Likos et al. 2005) | |
| KJ642617 | 3 | Nigeria | 1971 | (Nakazawa et al. 2015) | |
| MK783031 | 3 | A | Nigeria | 2017 | (Yinka-Ogunleye et al. 2019) |
| MK783029 | 3 | A | Nigeria | 2017 | (Yinka-Ogunleye et al. 2019) |
| MK783027 | 3 | A | Nigeria | 2017 | (Yinka-Ogunleye et al. 2019) |
| MK783028 | 3 | A | Nigeria | 2017 | (Yinka-Ogunleye et al. 2019) |
| MK783030 | 3 | A | Nigeria | 2017 | (Yinka-Ogunleye et al. 2019) |
| MK783033 | 3 | A | Nigeria | 2017 | (Yinka-Ogunleye et al. 2019) |
| MK783032 | 3 | A | Nigeria | 2017 | (Yinka-Ogunleye et al. 2019) |
| MT903339 | 3 | A | Nigeria | 2017 | (Mauldin et al. 2022) |
| MT903340 | 3 | A | Nigeria | 2018 | (Mauldin et al. 2022) |
| MT903338 | 3 | A | Nigeria | 2017 | (Mauldin et al. 2022) |
| MG693724 | 3 | A | Nigeria | 2017 | (Faye et al. 2018) |
| MN648051 | 3 | A.1 | Israel | 2018 | (Cohen-Gihon et al. 2020) |
| MT903342 | 3 | A.1 | Singapore | 2019 | (Mauldin et al. 2022) |
| MT250197 | 3 | A.1 | Singapore | 2019 | (Yong et al. 2020) |
| MT903341 | 3 | A.1 | Nigeria | 2018 | (Mauldin et al. 2022) |
| MT903343 | 3 | A.1 | UK | 2018 | (Mauldin et al. 2022) |
| MT903344 | 3 | A.1 | UK | 2018 | (Mauldin et al. 2022) |
| MT903345 | 3 | A.1 | UK | 2018 | (Mauldin et al. 2022) |
| ON676708 | 3 | A.1.1 | USA | 2021 | Gigante et al, 2022, CDC |
| ON585029-ON585036 | 3 | B.1 | Portugal | 2022 | (Isidro et al. 2022) |
| ON563414 | 3 | B.1 | USA | 2022 | (Gigante et al. 2022) |
| 3 | B.1 | Belgium | 2022 | (Vanmechelen et al. 2022) | |
| ON568298 | 3 | B.1 | Germany | 2022 | (Antwerpen et al. 2022) |
| ON674051 | 3 | A.2 | USA | 2022 | Gigante et al, 2022, CDC |
| ON676707 | 3 | A.2 | USA | 2021 | Gigante et al, 2022, CDC |
| ON675438 | 3 | A.2 | USA | 2022 | Gigante et al, 2022, CDC |