So, summer has come and gone, and it’s time for an updated whole genome molecular clock tree.
Compare this with the “Pre-Zibra phylogeographic summary” posted on Virological in May. The number of new sequences since then is not great. This may reflect the difficulties everyone has encountered in obtaining whole genomes directly from patient samples. Or it may be that many sequences have been generated but have not been shared.
The new genomes from French Polynesia in 2013-2014 strengthen the hypothesis of direct transmission from there to the Americas, and further highlight the need for much greater sampling in SE Asia. It is very possible that the 2016 cases in the Pacific (e.g. American Samoa) were not descended directly from FP, but are instead a separate introduction from a SE Asian reservoir. The Singapore outbreak was reported to have originated from this reservoir, not from the Americas.
Lots of spatial structure now evident. The common ancestor for the American lineage is still most likely to be in Brazil, but not definitively. Hopefully we’ll see some more 2014 genomes soon.
As before, Brazil = red, Pacific = blue, central America & Caribbean = orange, south America excl. Brazil = purple. Circle sizes at internal nodes are proportional to posterior probability. Analyses performed by Julien Theze. Credit for the sequences to the original data generators (see individual GenBank entries).
Yes, the number of complete genomes is a bit disappointing, but I am quietly hopeful that the flood gates are about to open in the next few days! It is good that several groups have had success with the new multiplex method.
Crappy indeed! With Nick and Josh’s protocol, however, we’re able to sequence samples with Ct’s of ~36. Still a few things to iron out, but I expect a lot more data over the next couple of months.
Hi Eddie, we’ve just finished a simulation study to look at the effects on phylogenetics of having varying number of partial American lineage genomes (ranging from 80% to 20% coverage) in the data set. Tree topology is reasonably robust if half of sequences have 60%-80% coverage. Lower coverage than that isn’t advisable. There are differences between clock and non-clock trees, presumably due to the effect of the priors. The clock rate is robust because a lot of the temporal signal comes from the older Asian/Pacific strains, and we only simulated partial coverage of American lineage strains. Will try to get something on bioRxiv soon.
I would be curious to hear from others in the community that have had success sequencing complete/near complete genomes from clinical samples by other methods other than PCR tiling schemes.
SE Asian sampling is pretty poor yes, we have a few more genomes to add to the mix from 2013 (n=2) and 2015 (n=1) from Thailand - I think they’ll be released next year, if not sooner, in Genbank. PLoS NTDs passed on the paper after review so I am revising and trying to get it out again.
Thanks Andrew! Virus Evolution…I hadn’t thought of that! Given the approval process our group (military) has to go through, studies need to go to journals. They don’t really ‘get’ the whole pre-print server thing, they’d rather just wait and send directly to a journal. I’ll look at virus evolution. My coauthors have also suggested JVI and AJTMH.
Yeah Nick and Josh’s protocol is a target-based approach (amplicons). We also have a SureSelect panel in the lab that we’re trying out and I have seen other institutions having some success with similar approaches. Generally speaking though, amplicon-based approaches appear to work better for most clinically relevant levels of virus (Ct ~35). In terms of approaches and based on a ‘standard’ qPCR assay:
Ct values:
< 28 - Use standard unbiased metagenomic approach (with rRNA depletion). HiSeq likely required.
28-32 - Use capture-based approaches (SureSelect, Hybrid Select, etc.). Could also use long amplicon-based approaches. HiSeq likely required.
32-37 - Use short amplicon-based approach (e.g. Primal from Josh/Nick, AmpliSeq, etc.). MiSeq sufficient.
37 and higher - Yeah… good luck.
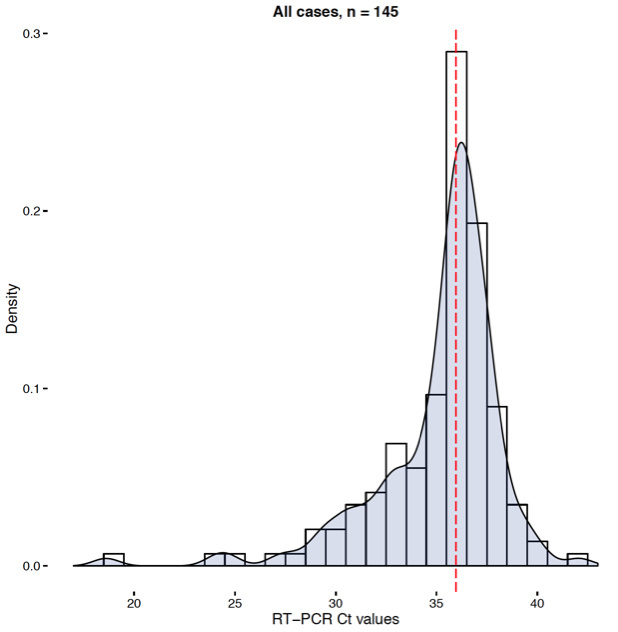
Most patient samples seem to be in the 34 - 38 range with only very few <32 and a fair number over 38. This is purely based on anecdotal evidence - I’m sure @n_j_loman could give you real data on that.
Sample selection? Saliva > Urine > Blood in our hands.
Not only are mosquitos going to have small amounts of virus, infected mosquitos are likely to be very rare. Would be good to know which species of mosquitos are carrying Zika and where (and when) but it wouldn’t be an efficient way of doing genomic surveillance. At least you can take samples from humans that you are know are infected. In my opinion, hospitalised patients is a good way of getting a representative sample because sick patients will likely have higher viremia, and the fact they are sick will be a property of the host not the virus. Sick patients will travel to the hospital meaning you can get reasonably good coverage of the area.
Interestingly not actually true - lots and lots of virus in mosquitos, so Nate is right. Three pools of mosquitos also tested positive in Miami, so not quite as rare as one might think.
I’m new to this whole arbovirus thing, but apparently having lots of virus in mosquitos is a common feature of these viruses (e.g. West Nile and others).