Towards Pandemic-Scale Ancestral Recombination Graphs of SARS-CoV-2
Shing H. Zhan1, Anastasia Ignatieva2,3,*, Yan Wong1,*, Katherine Eaton4, Benjamin Jeffery1, Duncan S. Palmer1, Carmen L. Murall4, Sarah P. Otto5, Jerome Kelleher1,†
1Big Data Institute , Li Ka Shing Centre for Health Information and Discovery, University of Oxford, United Kingdom
2Department of Statistics, University of Oxford, United Kingdom
3School of Mathematics and Statistics, University of Glasgow, United Kingdom
4National Microbiology Laboratory, Public Health Agency of Canada, Canada
5Department of Zoology and Biodiversity Research Centre, University of British Columbia, Canada
*Joint second author
†Correspondence: [email protected]
With increasing numbers of recombinant lineages emerging, it is crucial that methods for studying the evolution of SARS-CoV-2 fully integrate the process of viral recombination[1]. Here, we present an “alpha” version of an efficient method named sc2ts (optionally pronounced “scoots”) that simultaneously (i) detects recombinants at scale, (ii) reconstructs the evolutionary origins of SARS-CoV-2 recombinants, and (iii) integrates mutation and recombination cohesively into a network-like phylogeny, or ancestral recombination graph (ARG). Our method uses a standard HMM matching approach that has been previously used to construct large recombinant genealogies, and stores the results in the widely-used tskit “succinct tree sequence” format[2,3]. This enables an ARG of 1.27 million SARS-CoV-2 genomes to be stored in only 58 MB, loaded within 1 second, and analysed using Python / Jupyter notebooks on a typical laptop. We encourage others in the community to join us in improving this open-source project to create a robust computational platform for investigating the evolution and epidemiology of SARS-CoV-2 and beyond. Below, we highlight some of our key findings. For more details, please refer to our preprint[4].
Understanding the origins of recombinant lineages
Evaluating genomic evidence for new recombinants has until now been largely a manual process involving a variety of bioinformatics tools. Combining sc2ts results with the tskit library provides an easy way to examine the evolutionary context around the origins of SARS-CoV-2 recombinants. For example, users can visualise subgraphs within an ARG that shows putative origins of recombinant lineages, including their specific parent strains. Below is an example for the known Pango recombinants XA, XD, and XAG from the smaller of the two demonstration ARGs we have inferred (Figure 1). The sc2ts approach can detect recombination between very closely related lineages, as measured by how recently the parents of a recombinant shared a common ancestor (Figure 2).
Figure 1. Examples of Nextclade Pango X lineages (from the restricted ARG dated to June 2022). Genomic regions inherited at recombination events are shown; where samples are ancestral to further unplotted samples, the number of unplotted descendant samples is marked as “+1”, “+7”, etc. Note that the vertical position of nodes does not show absolute time, but relative rank (i.e. parents above children). For the identity of the sampled genomes in the plots above, as well as more complicated examples, please refer to our preprint.
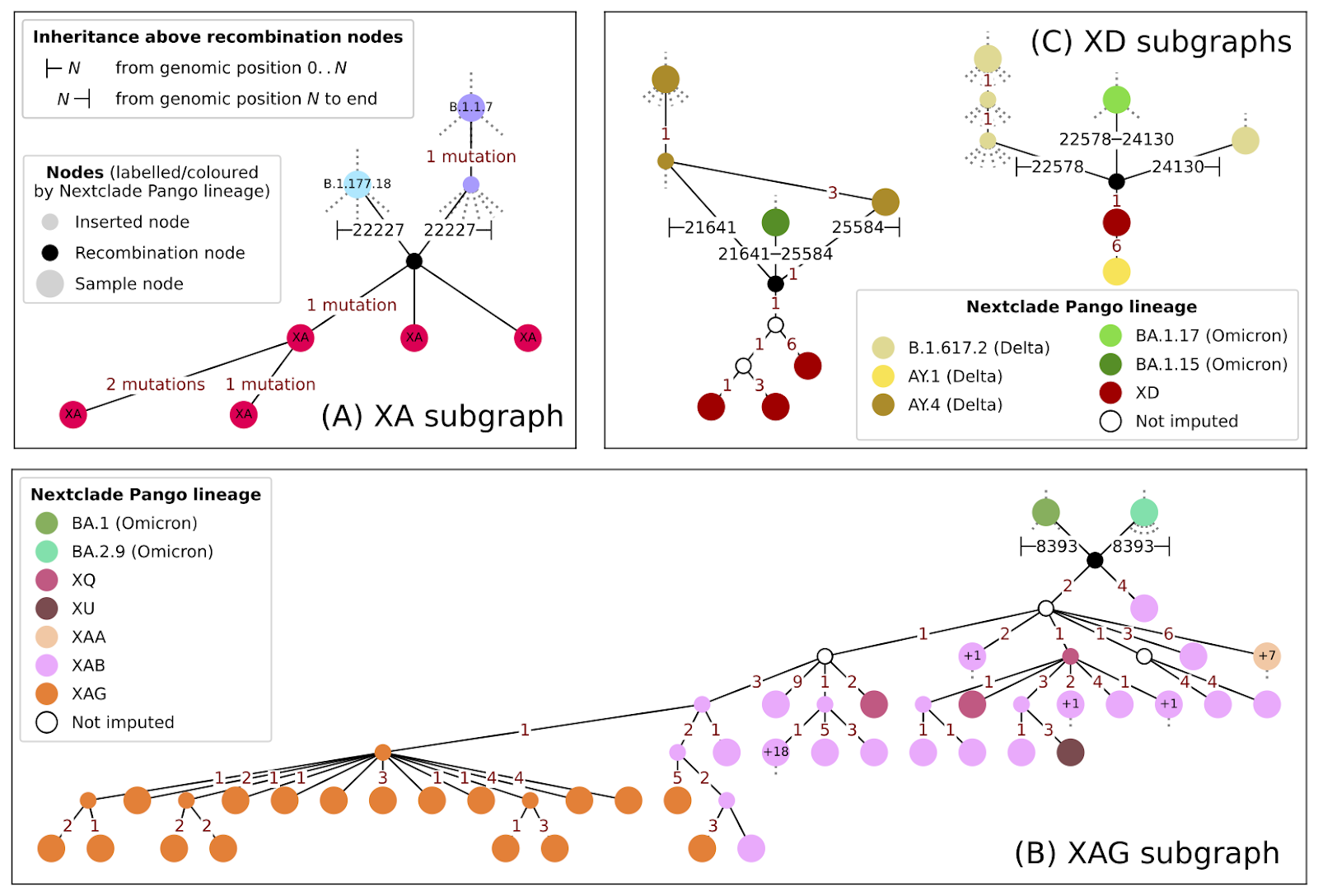
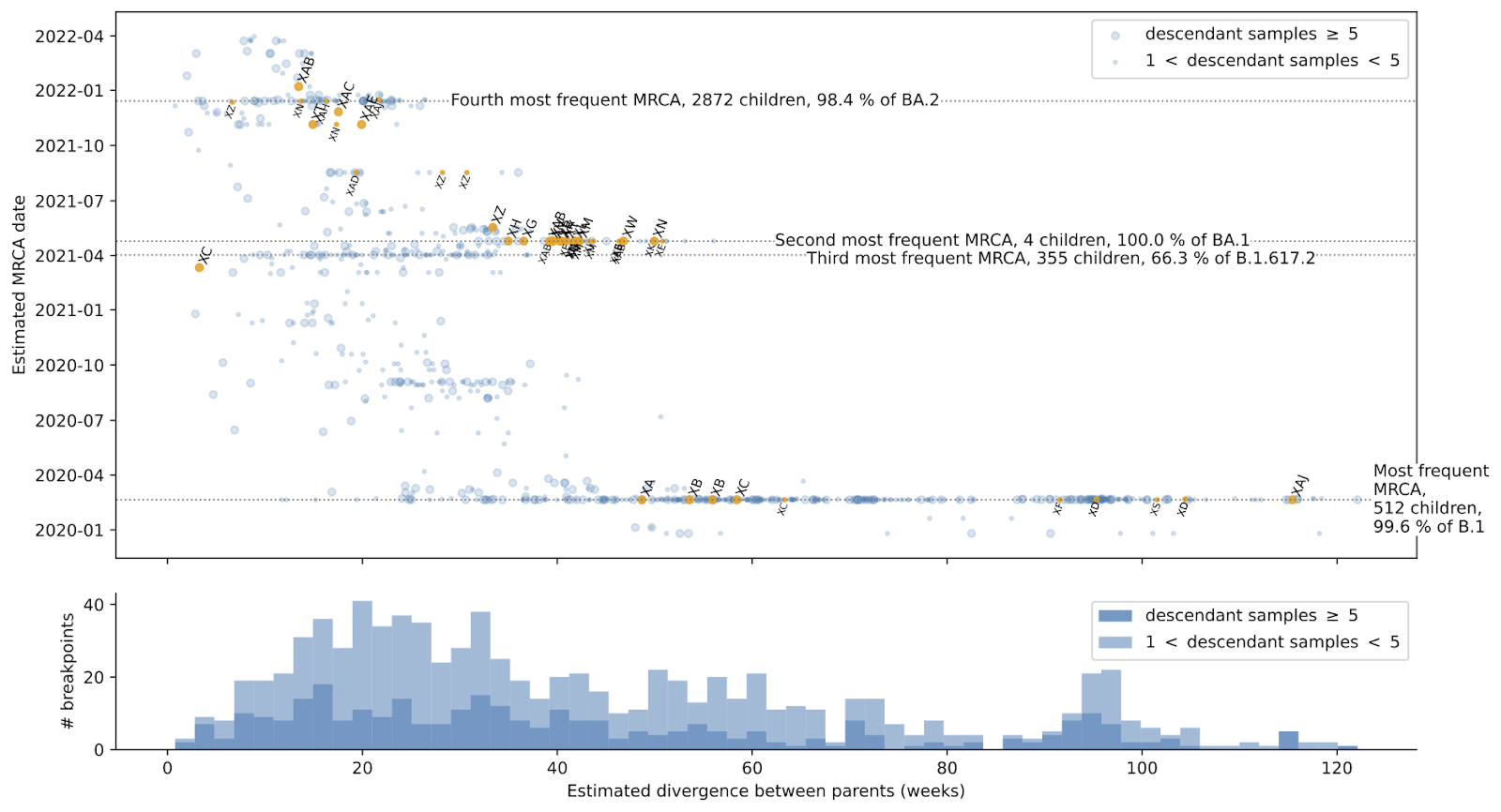
Figure 2. Dates of the most recent common ancestor (MRCA) between recombinant parents plotted against the divergence time between the parents (based on 763 putative recombination events from the restricted ARG dated to June 2022). MRCAs of parents associated with Pango-designated recombinants (XA, etc.) are identified in orange.
Building SARS-CoV-2 ARGs at scale
The key idea of our approach is to leverage information in the collection dates of SARS-CoV-2 samples. The method builds an ARG sequentially by adding batches of genomes one day at a time, moving forwards in time as the pandemic unfolds. Recombinant inheritance between new samples and their ancestors is inferred using a well-established HMM-based statistical approach. Local relationships among new samples are reconstructed using standard methods and parsimony-based refinements. Previous computational improvements in the scaling of the underlying HMM machinery[3] means that we expect to be able to apply sc2ts to most, if not all, reliably dated SARS-CoV-2 genomes.
As a demonstration of our approach, using relatively limited computational resources, we built two SARS-CoV-2 ARGs: one including all reliably dated samples up to June 2021, the other up to June 2022 but restricted to <1,000 samples per day. These contain 1.27M and 0.66M SARS-CoV-2 genomes, respectively (downloaded from GISAID). We encourage interested parties to contribute to further development of sc2ts before devoting additional computational resources to create larger ARGs of >10 million genomes.
Developing sc2ts further
Although the ARGs inferred by sc2ts closely agree with established patterns in the ancestry of SARS-CoV-2, we have identified some improvements that could be made to the inference process. The largest improvements are likely to come from correcting for issues with input data quality (in particular, improving the reliability of sampling times, filtering out poorly dated samples, or incorporating sampling time uncertainty into the method). We also identify suspected false positive recombinants, and these, together with known patterns of mutation and recombination, can be used to tune the HMM approach and its parametrization to maximise inference accuracy. Finally, improvements are possible in the parsimony-based refining steps that follow after HMM matching.
Code and data availability
The source code for sc2ts is available on GitHub:
The notebooks & code used for the analyses described here are in a separate repository:
The demonstration ARGs in tskit format, containing up to 1.27 million SARS-CoV-2 genomes, are subject to GISAID distribution conditions, hence we have not placed them online. They are available by request from the authors.
Genome data acknowledgements
We gratefully acknowledge all data contributors, i.e., the Authors and their Originating laboratories responsible for obtaining the specimens, and their Submitting laboratories for generating the genetic sequence and metadata and sharing via the GISAID Initiative, on which this research is based. Details of the GISAID data used are available at https://doi.org/10.55876/gis8.230329cd and included in the Supplemental Table at Towards Pandemic-Scale Ancestral Recombination Graphs of SARS-CoV-2 | bioRxiv.
References
- Neches RY, McGee MD, Kyrpides NC. 2020. Recombination Should Not Be an Afterthought. Nat. Rev. Microbiol. 18(11): 606–606.
- Kelleher J, Thornton KR, Ashander J, Ralph PL. 2018. Efficient Pedigree Recording for Fast Population Genetics Simulation. PLoS Comput. Biol. 14(11): e1006581.
- Kelleher J, Wong Y, Wohns AW, Fadil C, Albers PK, McVean G. 2019. Inferring Whole-Genome Histories in Large Population Datasets. Nat. Genet. 51(9): 1330–38.
- Zhan SH, Ignatieva A, Wong Y, Eaton K, Jeffery B, Palmer DS, Murall CL, Otto SP, Kelleher J. 2023. Towards Pandemic-Scale Ancestral Recombination Graphs of SARS-CoV-2 | bioRxiv