Darren Martin, Steven Weaver, Houryiah Tegally, Emmanuel James San, Eduan Wilkinson, Jennifer Giandhari, Sureshnee Naidoo, Yeshnee Pillay, Lavanya Singh, Richard J Lessells, Tulio de Oliveira, NGS-SA, Joel Wertheim, Philippe Lemey, Oscar MacClean, David Robertson, Ben Murrell, Sergei L Kosakovsky Pond
In late 2020, three relatively divergent SARS-CoV-2 lineages emerged in rapid succession: (i) B.1.1.7 or 501Y.V1 which will hereafter be referred to as V1 (6) (ii) B.1.351 or 501Y.V2 which will hereafter be referred to as V2 (7) and (iii) P.1 or 501Y.V3 which will hereafter be referred to as V3 (8). Viruses in each of the three lineages (which will hereafter be collectively referred to 501Y lineages) have multiple signature deletions and amino acid changing mutations (Figure 1), many of which impact key domains of the spike protein and five of which are shared between two or three the different lineages.
Figure 1. SARS-CoV-2 genome map indicating the locations and encoded amino acid changes of what we considered here to be signature mutations of V1, V2 and V3 sequences. Genes represented with light blue blocks encode non-structural proteins and genes in orange encode structural proteins:S encodes the spike protein, E the envelope protein, M the matrix protein, and N the nucleocapsid protein. Within the S gene, the receptor binding domain (RBD) is indicated by a darker shade and the site where the S protein is cleaved into two subunits during priming for receptor binding and cell entry is indicated by a dotted vertical line.
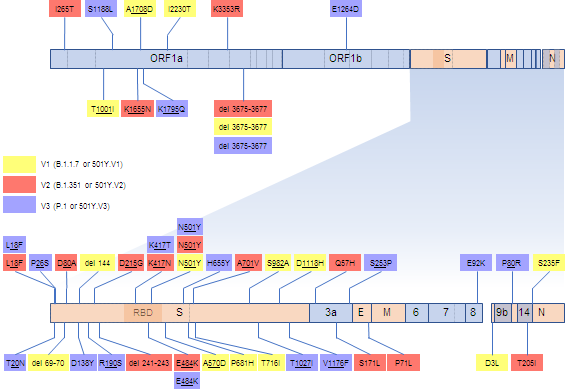
The simple existence of these five convergent mutations in different rapidly spreading viral lineages is extremely strong evidence that they each, either alone or in combination, provide some significant fitness advantage. The individual and collective fitness impacts of the other signature mutations in V1, V2 and V3 remain unclear. A key way to infer the fitness impacts of these mutations is to examine patterns of synonymous and non-synonymous substitutions at the codon sites where the mutations occurred. Specifically, it is expected that the most biologically important of these mutations will have occurred at codon sites that display substitution patterns that are dominated by non-synonymous mutations (i.e. mutations that alter encoded amino acid sequences); patterns that are indicative of positive selection.
Using a suite of phylogenetics-based natural selection analysis techniques, we examined patterns of positive selection within the protein coding sequences of viruses in the V1, V2 and V3 lineages to (1) determine the selective context within which these lineage emerged, (2) identify the mutations that have most likely contributed to the increased adaptation of these lineages, (3) test for ongoing adaptation, and (4) test whether this ongoing adaptation is associated with additional convergence between the lineages
There has been a recent qualitative shift in SARS-CoV-2 selective pressures
Analyses of positive selection on SARS-CoV-2 genomes undertaken prior to the emergence of 501Y lineages revealed patterns dominated by neutral evolution (2). There were, however, indications that some sites in the genome experienced episodes of positive selection (3–5). Through regular analyses of global GISAID data, starting in early 2020, we were able to track the extent and location of selective pressures on viral genomes. The power of these analyses to detect evidence of selection acting on individual codon sites progressively increased throughout 2020 with increasing numbers of sampled genome sequences and sequence diversification.
Even accounting for this expected increase in detection power, it is evident that a significant qualitative shift in selective pressures occurred ~11 months after SARS-CoV-2 cases were first reported in Wuhan city in December 2019. Specifically, during November 2020 this shift manifested in substantial increases in the numbers of SARS-CoV-2 codon sites that were detectably evolving under both positive and negative selection. This increase accelerated through January 2021, with sites found to be evolving under diversifying positive selection in S, N, and ORF3a (MEME or FEL p ≤ 0.0001) rapidly increasing in density from a baseline of between zero and four codons per Kb in November, to 10 or more codons per Kb at the beginning of 2021 (Figure 2A). This sudden increase in the numbers of sites that were detectably evolving under positive selection coincided with epidemic surges in multiple parts of the world in both hemispheres, some of which were driven by the emerging V1 and V2 lineages. The elevated density of sites under selection in S, N and ORF3a in January is also apparent when 501Y sequences are excluded from the dataset used for the January 2021 analysis. This indicates that the emerging 501Y lineages alone are unlikely to be the cause of this selective shift.
Among the 37 signature mutation sites in V1, V2 and V3 (Figure 1), 14 were detectably evolving under positive selection prior to December 2020 whereas this number increased to 31 by January 2021 (Figure 2C). The only signature mutation shared between any of the three 501Y lineages that was detectably evolving under positive selection before December 2020 was S/18 which was weakly detected for the first time in October.
Our regular tracking of positively selected SARS-CoV-2 codon sites Before November 2020 therefore yielded no clear indications that non-synonymous substitutions at the crucial RBD sites S/417, S/484 or S/501 (the other key convergent signature mutation sites in the 501Y lineages) might provide SARS-CoV-2 with any substantial fitness advantages. Instead, the sporadic weak selection signals that these analyses yielded between July and November were of adaptive amino acid substitutions in the Spike N-terminal domain (S/18, and the V3 signature site, S/26), near the furin cleavage site (the V3 signature site, S/655), and in C-terminal domain (the V3 signature site, S/1027, and the V1 signature site, S/1118). Conversely, for much of the latter half of 2020 the relatively strong and consistently detected selection signals at the V1 signature site, N/3, and the V2 signature sites, ORF1ab/265 (nsp2 codon 85) and ORF3a/57, clearly indicated that some substrituions at these sites were likely adaptive.
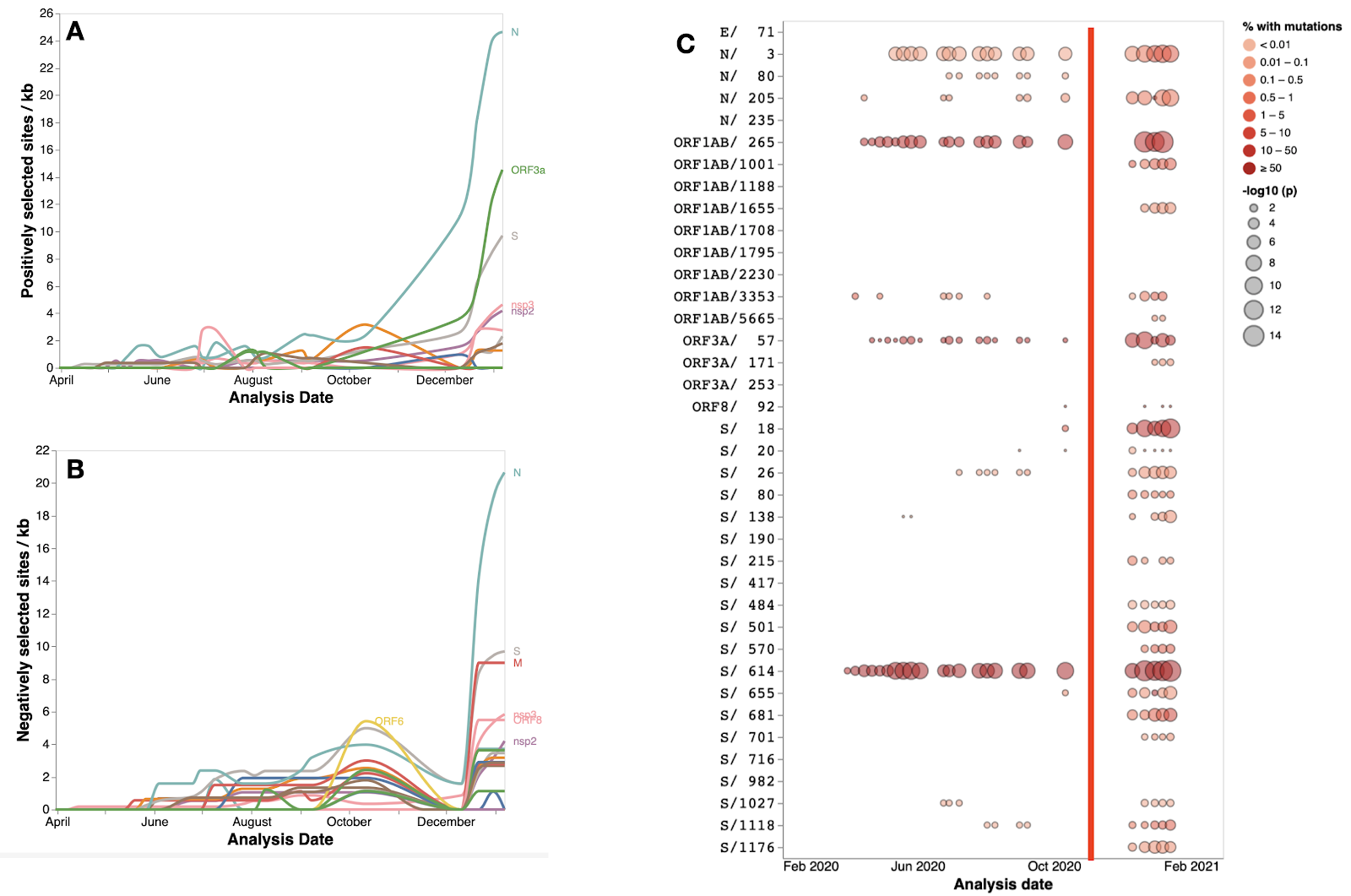
Figure 2. Signals of positive and negative selection at individual codon sites that were detectable with either the MEME or FEL methods at different times between March 2020 and January 2021. A/B. The gene-by-gene per Kb density of codons detectably evolving under positive/negative selection between March 2020 and January 2021. C Signals of positive selection detected at 37 V1, V2 and V3 signature mutation sites between March 2020 and January 2021. Also included for reference is S/614, the site of the D614G mutation that is present in all three of the 501Y lineages. For the circles the intensity of red indicates the proportion of analysed sequences that have mutations at a given site and the size of the circle indicates the statistical significance of the MEME and IFEL positive selection tests. The vertical red line indicates the 1st November 2020.
Taken together, these patterns of detectable selection suggest that the selective value of RBD signature mutations may have only manifested after a selective shift that occurred shortly before November.
Signals of ongoing selection within the V1, V2 and V3 lineages
We collected all sequences assigned to B.1.1.7 (V1), B.1.351 (V2), and P1 (V3) PANGO lineages (28) in GISAID as of Feb 02, 2021, and subjected them to a comprehensive battery of gene- and codon-level selection tests (see Methods).
We first tested the 501Y lineage sequences to determine whether, at the level of individual genes, they were evolving under quantitatively different selective pressures than other SARS-CoV-2 lineages. To each of the individual V1, V2, and V3 gene datasets we added algorithmically selected non-501Y lineage reference sequences that were representative of historical SARS-CoV-2 diversity (sampled prior to Oct 15th, 2020). We then analysed each of these datasets to compare the selective processes operating on the reference sequences relative to the V1, V2 and V3 lineage sequences since their emergence. The relative lack of sequence divergence in the analysed datasets meant that the power of these gene-wide analyses was expected to be low, especially for V3 with few sequences available. However, among the genes with sufficient clade-level phylogenetic resolution, using BUSTED[S] (29), we found evidence of episodic diversifying selection in the N gene (all clades), nsp3 (V1/V2), S (V3), and RdRp, endornase, exonuclease, ORF7 and ORF8 in V1. Though, it should be noted that the gene-wide signal of positive selection detected in the V1 ORF8 is likely a consequence of a mutation that introduced a stop codon into this ORF at position 27.
We next examined the different lineage-specific datasets with background references for evidence of positive selection at individual codon sites using MEME (30) and IFEL (31). These analyses revealed evidence of positive selection at 111 individual codon sites across all lineages including 43 in V1, 64 in V2, and 14 in V3 (Table S1; Combined view of sites under selection in N501Y clades / Sergei Pond / Observable). This is indicative of substantial ongoing adaptation of V1 and V2 sequences since the emergence of these lineages. V3 sequences might also be adaptively evolving but there is too little sequence data currently available to convincingly detect selection at individual codon sites in this lineage.
Signals of ongoing mutational convergence at 501Y lineage signature mutation sites
Notable among the lineage-specific positive selection signals detected at 111 individual codon sites of 501Y lineage sequences were those detected at 21 of their signature mutation sites (6/11 of the V1 signature sites, 8/14 of the V2 ones and 12/17 of the V3 ones; see underlined codon site numbers in Figure 1). Given both that each lineage was defined by the signature mutations along the phylogenetic tree branch basal to its clade, and that these basal branches were included in the lineage specific selection analyses, these selection analysis results were biased in favour of detecting the signature mutations as evolving under positive selection. We therefore used the selection results for signature mutation sites only to identify the signature mutations that, relative to the background reference sequences, were evolving under the strongest degrees of positive selection.
Most noteworthy of the signature mutation sites that displayed the strongest evidence of lineage-specific positive selection are codons S/18, S/80, S/417, S/484, S/501, and S/701 in that all are either suspected or known to harbour mutations with potentially significant fitness impacts (9,10,21,23,24,27,36).
There are particularly interesting ongoing mutational dynamics at codons S/18 and S/484 in the V1 lineage. Although neither of these codons are signature mutation sites in the V1 lineage, in multiple independent instances mutations have occurred at these sites that converge on signature mutations seen in the V2 and V3 lineages.
Independent L18F amino acid changes are seen twice in the V1 lineage and at least four times in the V2 lineage whereas all currently sampled V3 lineage sequences have this mutation. S/18 falls within multiple different predicted CTL epitopes (37) and the L18F mutation is known to reduce viral sensitivity to some neutralizing monoclonal antibodies (27). An F at residue S/18 is also observed in 10% of other known Sarbecoviruses and the L18F mutation is the tenth most common in currently sampled SARS-CoV-2 genomes. Having occurred independently numerous times since the start of the pandemic, S/18 is also detectably evolving under positive selection in the global SARS-CoV-2 genome dataset (MEME p-value = 6.9 x 10^-13).
More concerning than these convergent L18F mutations are the convergent E484K mutations that have independently arisen at least twice within the V1 lineage and are arising independently elsewhere. These mutations converge on a significant signature mutation found in the V2 and V3 lineages that decreases viral sensitivity to both infection- and vaccine-induced neutralizing antibodies (17,22–24,36) and, in conjunction with the N501Y mutation, increases the affinity of Spike for human ACE2 receptor sites (10,11).
Similar convergence patterns to those observed at S/18 and S/484 can be seen at seven other signature mutation sites: S/26, S/681, S/701, S/716, S/1176, and N/80. Of these S/681, S/701, and S/716 fall within 30 residues of the biologically important furin cleavage site (S/680 to S/689). Whereas some V2 sequences have independently acquired the signature V1 mutations, P681H and T716I, some V1 sequences have independently acquired the V2 signature mutation, A701V. Any of these three mutations might directly impact the priming of spike for ACE2 binding and, consequently, the efficiency of viral entry into host cells (38). SARS-CoV-2 variants with deletions of the furin cleavage site have reduced pathogenicity (39,40) and, although presently untested, it is plausible that the P681H mutation - which falls within this site, might increase the efficiency of furin cleavage by replacing a less favourable uncharged amino acid with a more favourable positively charged basic one (38). Whereas S/716 is not detectably under selection in either the lineage-specific or global SARS-CoV-2 datasets, sites S/681 and S/701 are both detectably evolving under positive selection in the global SARS-CoV-2 dataset (MEME p-values = 1.6 x 10^-6 and 7.9 x10^-3 respectively): important indicators that are consistent with the P681H and A701V mutations being adaptive.
Non-convergent mutations at signature mutation sites might still be evolutionarily convergent
In addition to the ten positively selected signature mutation sites displaying evidence of convergent mutations between the V1, V2 and V3 lineages, five positively selected signature mutation sites (S/20, S/80, S/138, S/190 and S/215) display evidence of only non-convergent mutations, where the same site is mutated as in another lineage, but to a different amino acid. All five of these codon sites are detectably evolving under positive selection in both individual lineage specific datasets and, with the exception of S/190, the global SARS-CoV-2 dataset (Figure 3).
Paradoxically, the diverging mutations at these five sites might in fact also be contributing to the overall patterns of evolutionary convergence between the lineages; just via different routes.The N-terminal domain of spike where all five of these sites fall, (and S/80 in particular) is targeted by some monoclonal and infection-induced neutralizing antibodies (27) and it is therefore possible that these sites could be evolving under immunity-driven diversifying selective pressures. Therefore while mutations at S/20, S/80, S/138, S/190 and S/215 in different lineages do not converge on the same encoded amino acid states, they might nevertheless still be convergent on similar fitness objectives: such as is likely the case with the also not-strictly-convergent V2 K417N and V3 K417T signature mutations.
Positive selection may be driving further convergence at non-signature mutation sites
In addition to lineage-specific signals of positive selection being detected at 21 of the signature mutation sites that characterize each of V1, V2 and V3 (Figure 1), such signals were also detected at 90 non-signature mutation sites (Supplementary Table 1). To test whether positive selection acting at these codon sites might be favouring convergent amino acid changes across the three lineages we examined each of the 90 non-signature mutation sites that were detectably evolving under positive selection in any one of the lineages for evidence of convergent mutations having occurred in either of the other two lineages. This revealed the occurrence of convergent mutations between sequences in different lineages at 26/90 (28.9%) of these sites including eleven in ORF1a and twelve in the S-gene (Figure 3).
Most notable among the mutations occurring at these non-signature sites are spike L5F mutations seen in V1 and V2 sequences. S/5 has been detectably evolving under positive selection in the global SARS-CoV-2 dataset since May 2020 (MEME p-value = 1.6 x 10^-16) (3) and is detectably evolving under positive selection in both the V1 (p=0.08, marginal evidence) and V2 lineages (p=0.001). L5F mutations have arisen independently at least eight times in the V1 lineage and at least twice in the V2 lineage. The precise biological significance of the L5F mutation remains unknown but it falls within CTL epitopes that are predicted to be recognizable by a broad array of different HLA types (Campbell et al., 2020) and is therefore potentially a CTL escape mutation.
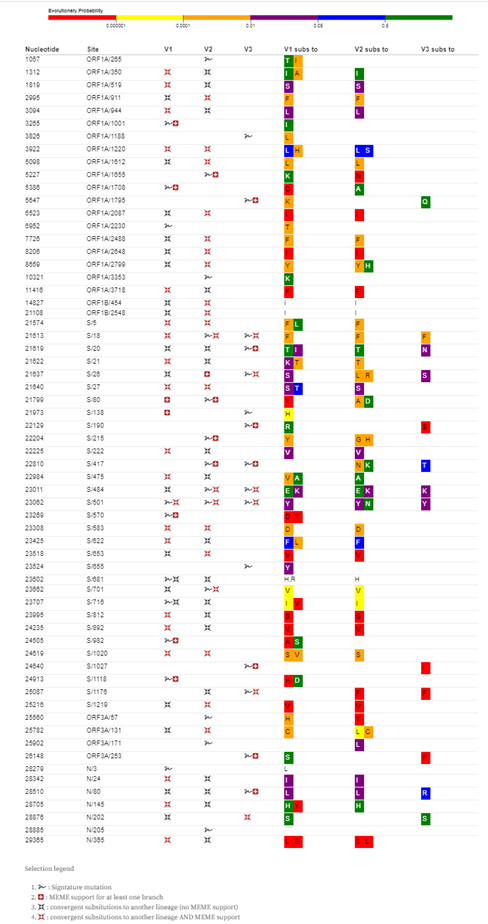
Figure 3. Genome sites where signature and convergent mutations occur within the V1, V2 and V3 lineage sequences. Labels within the coloured blocks indicate amino acid substitutions occurring at individual sites whereas the colours of the blocks indicate model-based predictions of the probable evolutionary viability of the observed amino acid substitutions based on the numbers of times these substitutions have been observed in related coronaviruses that infect other host species. The absence of colour indicates unprecedented substitutions, red indicates highly unusual substitutions and green indicates common substitutions seen at homologous sites in non-SARS-CoV-2 coronaviruses.
Collectively, convergent amino acid changes in individual clades of the V1 or V2 lineages respectively account for 18/256 and 17/112 of all the non-synonymous mutations mapping to internal branches of these clades. This is significantly higher (permutation p-val < 1 x 10^-6) than the total number of expected internal branch convergent mutations (approximately one for both V1 and V2) under absolute neutrality and uniform mutation rates. All 26 genome sites where convergent mutations occur are detectably evolving under positive selection in at least one of the V1, V2 or V3 lineages. Further, 20/26 of the codons where these convergent non-signature site mutations occur are also detectably evolving under either pervasive or episodic positive selection in the global SARS-CoV-2 dataset (MEME p-values < 0.05). Collectively this is strong evidence that an appreciable proportion of the convergent non-signature site mutations are adaptive.
Conclusion
We find evidence that the emergence of the 501Y lineages coincided with a marked global change in the selective environment within which SARS-CoV-2 is evolving. Against this backdrop the 501Y lineages all display evidence of substantial ongoing adaptation that, besides in many cases involving mutations that converge on the signature mutations of other 501Y lineages, also involve multiple other convergent mutations at non-signature sites: a pattern which suggests both that viruses in all three lineages may be climbing very similar adaptive peaks, and, therefore, that viruses in all three lineages are likely in the process of converging on a similar adaptive end-point.
Methods
Unless specified otherwise, all analyses were performed on a single gene (e.g. S) or peptide product (e.g. nsp3); since genes/peptides are the targets of selection.
We aligned all sequences from an individual lineage (i.e. V1, V2 or V3) and reference (GISAID unique haplotypes in Spike) sequences to the GenBank reference genome protein sequence for the corresponding segment using the codon-aware alignment tool, bealign (part of the BioExt Python package, also used by HIV-TRACE (54) with the HIV-BETWEEN-F scoring matrix (similar viral sequences). This approach did not consider insertions relative to the reference genome in subsequent analyses. Deletions were treated as missing data and were not specifically tested for with codon models. No convincing signals of recombination were detected in any of the alignments using RDP5 (55).
Because the codon-based selection analyses that we performed gain no power from including identical sequences, and minimal power from the inclusion of sequences that are essentially identical, we filtered V1, V2, V3, and reference (GISAID) sequences using pairwise genetic distances complete linkage clustering with the tn93-cluster tool. All groups of sequences that are within D genetic distance (Tamura-Nei 93) of every other sequence in the group and represented by a single (randomly chosen) sequence in the group. We set D at 0.0001 for lineage-specific sequence sets, and at 0.0015 for GISAID reference (or “background”) sequence sets. We restricted the reference set of sequences to those sampled before Oct 15th, 2020.
We inferred a maximum likelihood tree from the combined sequence dataset using raxml-ng using default settings (GTR+G model, 20 starting trees). We partitioned internal branches in the resulting tree into two non-overlapping sets used for testing and annotated the Newick tree. Because of lack of phylogenetic resolution in some of the segments/genes, not all analyses were possible for all segments/genes. In particular this is true when lineage V1, V2 or V3 sequences were not monophyletic in a specific region, and no internal branches could be labeled as belonging to the focal lineage.
We used HyPhy v2.5.27 (56) to perform a series of selection analyses. Analyses in this setting need to account for a well-known feature of viral evolution (41) where terminal branches include “dead-end” (maladaptive or deleterious on the population level) (57) mutation events within individual hosts which have not been “seen” by natural selection, whereas internal branches must include at least one transmission event. However, because our tree is reduced to only include unique haplotypes, even leaf nodes could represent “transmission” events, if the same haplotype was sampled more than once (and the vast majority were). We performed:
- Gene-level tests for selection on the internal branches of the V1,V2 or V3 clades using BUSTED (29) with synonymous rate variation enabled.
- Codon site-level tests for episodic diversifying (MEME) (29) and pervasive positive or negative selection (FEL) (59) on the internal branches of the V1,V2 or V3 clades.
.