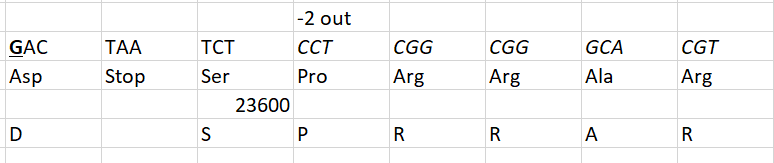
How do you explain TAA? And what are the chances that all 19 mutations would be in the middle nucleotide of the codon?