Spike protein sequences of Cambodian, Thai and Japanese bat sarbecoviruses provide insights into the natural evolution of the Receptor Binding Domain and S1/S2 cleavage site
Edward C. Holmes1, Kristian G. Andersen2,3, Andrew Rambaut4 and Robert F. Garry5,6,*
1Marie Bashir Institute for Infectious Diseases and Biosecurity, School of Life and Environmental Sciences and School of Medical Sciences, The University of Sydney, Sydney, Australia.
2Department of Immunology and Microbiology, The Scripps Research Institute, La Jolla, CA 92037, USA.
3Scripps Research Translational Institute, La Jolla, CA 92037, USA.
4Institute of Evolutionary Biology, University of Edinburgh, Edinburgh, UK.
5Department of Microbiology and Immunology, Tulane University Medical Center, 1430 Tulane Avenue, New Orleans, Louisiana 70112 USA.
6Zalgen Labs, LLC, Germantown, MD, USA.
*corresponding author: E-mail: [email protected].
Introduction
SARS-CoV and SARS-CoV-2 are members of the Sarbecovirus subgenus of betacoronaviruses (family Coronaviridae). Two sarbecoviruses lineages, GX (Guangxi) and GD (Guangdong), have been identified in Malayan pangolins, Manis javanica, illegally imported into China (Liu, Chen and Chen, 2019; Lam et al., 2020). A variety of sarbecoviruses have also been described in bats of the genus Rhinolophus, although it is evident that the diversity of this subgenus in wildlife species has been greatly under-sampled (Boni et al., 2020). While close relatives of SARS-CoV have been identified in bats, civets and other animals (Li, 2008), the immediate progenitor of SARS-CoV-2 is unknown (Andersen et al., 2020). The bat coronavirus RaTG13 genome was sequenced from a sample from a Rhinolophus affinis captured at Mojiang cave in Yunnan province, China in 2013 (Ge et al., 2016; Zhou P et al., 2020a,b). RaTG13 remains the virus with the highest overall sequence similarity to SARS-CoV-2, although because of frequent recombination patterns of sequence similarity vary across the genome. Recently, new sarbecoviruses have been sequenced from bats sampled in Cambodia, Thailand and Japan (Hul et al., 2021, Wacharapluesadee et al., 2021; Murakami et al., 2020). Here, we reveal more of the natural evolution of sarbecoviruses by analyzing the spike protein sequences of these viruses. We also discuss some implications of these new sequences for understanding the proximal origin of SARS-CoV-2.
Methods
Spike protein sequences from a variety of sarbecoviruses were selected for comparative analysis as described in Table 1.

All spike amino acid sequences from these viruses were aligned using Clustal Omega (Sievers et al., 2011) and adjusted by visual inspection.

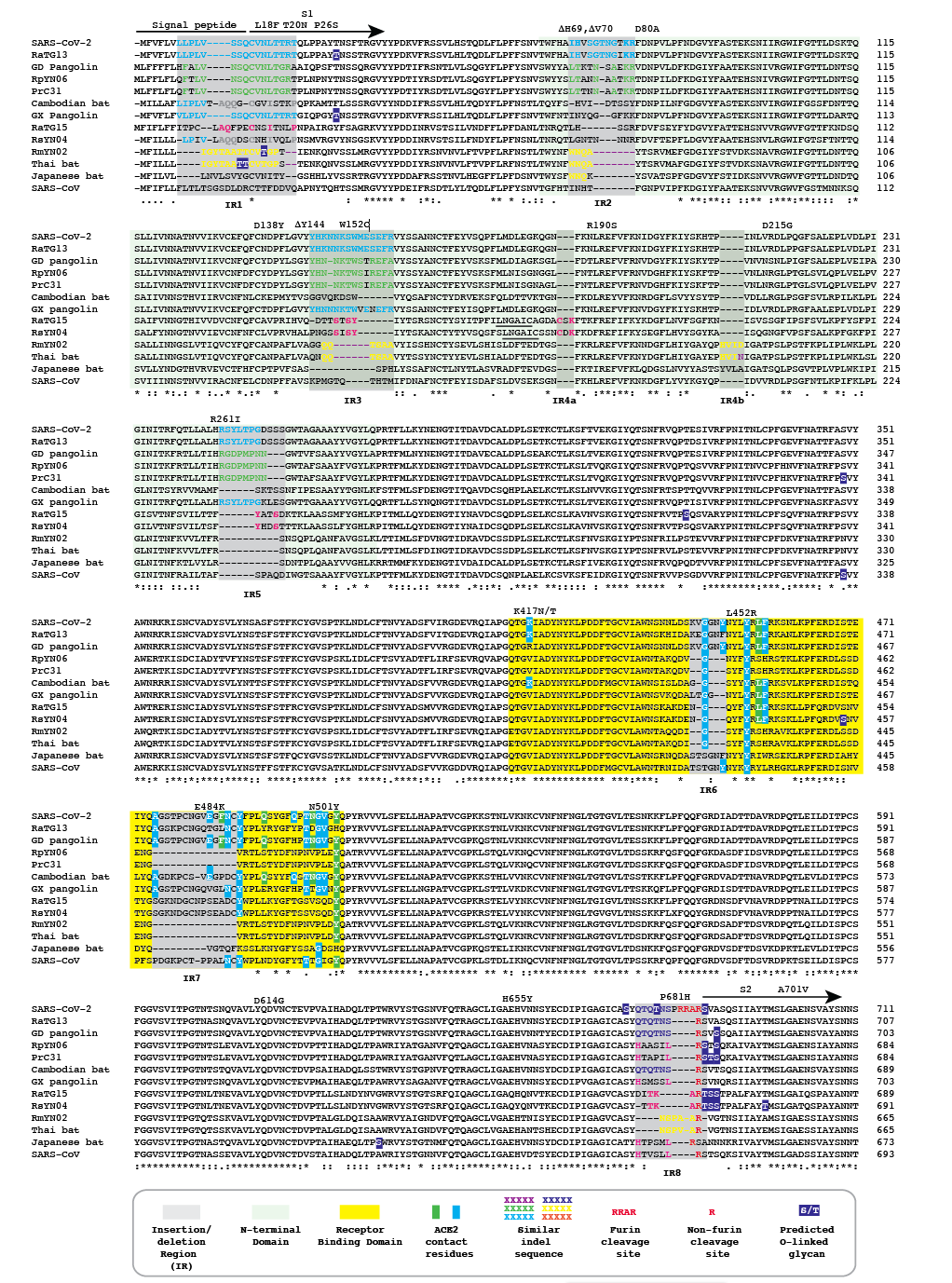
Figure 1 | Amino acid alignment of the spike protein sequences of Cambodian, Thai and Japanese bat coronaviruses and other sarbecoviruses. The signal peptide, S1 subunit and part of S2 are shown. Positions of selected substitutions or deletions in ‘variants of concern’ are indicated.
Results
We refer to regions of the spike protein that have acquired insertions or deletions during sarbecovirus evolution as “Indel Regions” (Garry et al., 2021). Our comparative analysis identified 8 Indel Regions, covering amino acid positions 7-22, 68-78, 145-158, 246-255, 444-449, 475-489, 675-686, as well as a 4 amino acid insertion between positions 230 and 231, relative to the SARS-CoV-2 genome (Figs 1 and 2). Notably, RaTG13, GX pangolin coronavirus and SARS-CoV-2 share nearly identical sequences in Indel Regions 1, 3 and 5 located in the N-terminal portions of the spike protein (Fig. 1, blue letters, Fig. 2). In contrast, Indel Regions 1, 3 and 5 are divergent in GD pangolin coronaviruses, while Indel Regions 7 and 8 of GD pangolin coronavirus are more similar to SARS-CoV-2 (Indel Region 7: 13 of 13 amino acids, Indel Region 8: 8 of 12 amino acids) than those of GX pangolin coronavirus (Indel Region 7: 10 of 13 amino acids, Indel Region 8: 2 of 12 amino acids).
Cambodian bat coronavirus spike differs from other sarbecoviruses in Indel Regions 1, 2, 3 and 5. Indel Region 7, which includes part of the RBD, of the Cambodian bat coronavirus spike protein is similar to Indel Region 7 of RaTG13 (9 of 13 amino acids), GD pangolin coronavirus (9 of 13 amino acids) and SARS-CoV-2 (10 of 13 amino acids). Indel Region 8 includes the S1/S2 junction of the spike protein. Indel 8 of Cambodian bat coronavirus is identical to that of RaTG13 (8 of 8 amino acids) and GD pangolin coronavirus (8 of 8 amino acids), and aside from the lack of the polybasic cleavage site (PRRA) insertion at the S1/S2 junction, it is also homologous to Indel Region 8 of SARS-CoV-2 (8 of 12 amino acids).
The spike proteins of RmYN02 and Thai bat coronavirus viruses share identical or highly similar sequences in every Indel Region (Fig. 1, purple letters, Fig. 2), with 98.0% amino acid identity (99.3% similar amino acids) overall, reflecting their close phylogenetic relationship across the complete genome.
The Japanese bat coronavirus spike protein sequence has several similarities to the Thai bat coronavirus and RmYN02 spike sequences. This includes similar-sized insertions or deletions at Indel Regions 1, 2, 4 and 5. Notably, these are the only viruses that have a 4 amino acid insert in Indel Region 4 relative to the other sarbecovirus spikes analyzed. The Japanese bat coronavirus spike protein has a sequence in Indel Region 6 that is longer than those in the newly described isolates from Thailand and Cambodia. The spike protein sequence of the Japanese bat coronavirus also differs from the other sarbecovirus spikes analyzed at Indel Region 8, containing a one amino acid insertion relative to other sarbecovirues following the S1/S2 cleavage site.
The RaTG13 spike protein shares 97.4% amino acid identity and 98.7% nucleotide sequence similarity with the SARS-CoV-2 spike protein. Although the GD pangolin coronavirus spike has less overall sequence similarity to SARS-CoV-2 spike (89.8% identical, 96.0% similar), it exhibits a larger number of predicted contact residues with ACE2 than RaTG13 (Lam et al., 2020; Andersen et al., 2020). Specifically, the spike protein sequence of the GD pangolin shares 5 of 6 predicted ACE2 contact residues with SARS-CoV-2, compared with only 1of 6 for the RaTG13 spike protein. Here, we update these analyses using structural studies that reveal a more extensive interaction of SARS-CoV-2 spike with ACE2 than predicted by computational modelling (Lan et al., 2020; Shang et al., 2020). Specifically, the GD pangolin coronavirus shares 15 of 18 putative contact residues that SARS-CoV-2 spike protein makes with ACE2 (Figs. 1 and 2). The spike protein of the Cambodian bat coronavirus shares a different set of 15 ACE2 contact residues with SARS-CoV-2 spike. In contrast, RaTG13 and GX pangolin coronavirus share 11 of 18 and 10 of 18 contact residues, respectively.
Notably, SARS-CoV-2, RaTG13, GD pangolin coronavirus and the Cambodian bat coronavirus contain a QTQTNS motif adjacent to Indel Region 8 (Fig. 1, blue letters, Fig. 2). In the SARS-CoV-2 spike protein, the QTQTNS motif directly precedes the furin cleavage site (Indel region 8). This concentration of polar amino acids may provide a favorable landing site for furin and other proteases (Tian, 2009). RmYN02 is notable for its partial similarity at the S1/S2 junction to SARS-CoV-2 spike (Zhou H et al., 2020). The RmYN02 spike protein has the sequence NSPVAR in indel 8, which is similar to the furin cleavage site NSPRRAR found in SARS-CoV-2 (Fig. 1, orange lettering, Fig. 2). A similar, but not identical, sequence NSPAAR is present in the Thai bat coronavirus. This motif can be depicted as NSPXX/-AR.
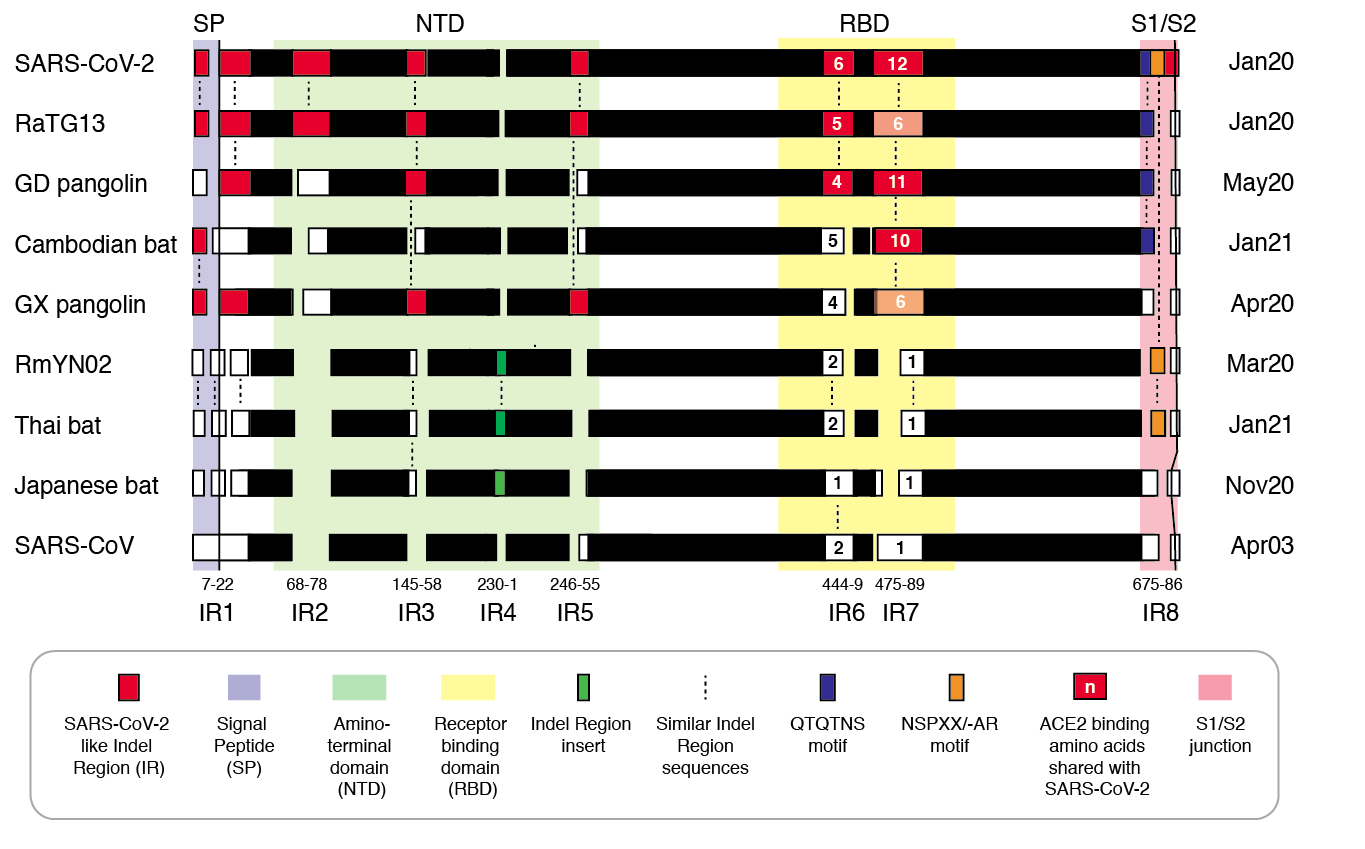
Figure 2 | Insertion/deletion structure of the Cambodian, Thai and Japanese bat coronavirus spike protein sequence compared to other sarbecoviruses. Amino acids delineating indel regions are numbered according to the SARS-CoV-2 spike sequence. Indel lengths are not drawn to scale. Numbers of amino acids in common with SARS-CoV-2 RBD In Indel Regions 6 and 7 include surrounding sequences. Month/Year of sequence deposit is indicated.
Discussion
The RBD of the Cambodian bat coronavirus spike is similar to the RBDs of GD pangolin coronavirus (14 of 18 ACE2 contact residues shared) and SARS-CoV-2 (15 of 18 ACE2 contact residues shared). The Cambodian bat coronavirus was detected in a Rhinolophus shameli captured in 2010, but only sequenced in recent months (Hul et al., 2021). The virus was sequenced by a research group that is independent from those that first generated and analyzed the sequences of pangolin coronaviruses (Liu P, Chen and Chen, 2019; Lam et al., 2020; Xiao et al., 2020) and SARS-CoV-2 (Wu L et al., 2020; Zhou P et al., 2020a,b). The repeated independent detection of viruses from different animal species with highly similar RBDs indicates that these viruses arose naturally (Boni et al., 2020).
The RBD of the Cambodian bat coronavirus provides a divergent example of a sequence that binds ACE2. The binding specificity of Cambodian bat coronavirus RBD remains to be determined. In this regard, there is no evidence that the ACE2 binding solution that SARS-CoV-2 shares in part with Cambodian bat coronavirus as well as RaTG13 and GD pangolin coronavirus is specific for human ACE2. On the contrary, SARS-CoV-2 binds efficiently to ACE2 of several animal species (Wu F et al., 2020; Shang et al., 2020), thereby invalidating claims that the SARS-CoV-2 RBD was either selected or specifically optimized for human ACE2 binding (Zhan, Deverman, and Chan, 2020; Piplai et al., 2020). Further evidence that the SARS-CoV-2 RBD is not specifically adapted to human ACE2 is provided by repeated examples of human-to-animal transfers that require few, if any, RBD mutations (Garry, 2021). Moreover, the RBD is the site of several mutations in newly detected SARS-CoV-2 variants: this suggests that the human ACE2 binding is not optimal and is still subject to adaptive evolution as the virus spreads through the human population (Rambaut et al., 2020; Tegally et al., 2020; Faria et al., 2022).
The RmYN02 genome was sequenced from a Rhinolophus malayanus sampled in Yunnan province China in June 2019 (Zhou H et al., 2020). The Thai bat coronavirus genome was sequenced from a Rhinolophus acuminatus sampled by an independent research group one year later (Wacharapluesadee al., 2021). The high similarity of the newly-derived spike sequence of Thai bat coronaviruses with RmYN02 spike (98.0% identity, 99.3% similar amino acids over a 1227 amino acid overlap) shows that RmYN02 cannot be a contrived or manipulated virus.
Furin cleavage sites have been noted at the S1/S2 junctions in members of four betacoronavirus subgenuses, and while not universally present, can also be found in other human coronaviruses (Wu and Zhao, 2020). The newly determined S1/S2 junction sequences of Thai, Japanese and Cambodian bat coronaviruses spikes add to evidence that this region of the spike protein represents an evolutionary “hotspot”. Notably, the QTQTNS motif near the S1/S2 cleavage site is present in Cambodian bat coronavirus, RaTG13, GD Pangolin coronavirus and SARS-CoV-2. None of these sequences were determined until after the COVID-19 pandemic began. Likewise, the NSPXX/-AR motif in Thai bat virus, RmYN02 and SARS-CoV-2 spike had not been detected in any coronavirus sequenced prior to 2019. Japan bat coronavirus also has a distinct S1/S2 junction, and provides another example of an apparent insertion near the site. These observations provide further strong evidence for the evolutionary volatility of the S1/S2 cleavage site and that the furin cleavage site arose in SARS-CoV-2 via a natural insertion process (Zhou H et al. 2020).
The new sequences of sarbecoviruses from bats captured in Cambodia, Thailand and Japan fill important gaps in the evolutionary history of the sarbecoviruses. Recently, an additional sarbecovirus has been sampled from a Manis pentadactyla (Chinese pangolin) collected in 2017 in Yunnan province, China (GISAID ID EPI_ISL_610156) (Li et al., 2021). This new independently-derived sequence from a different pangolin species provides strong confirmation that the original pangolin coronavirus sequences were genuine and accurate despite some supposition to the contrary (Chan and Zhan, 2020). In addition, Wacharapluesadee and co-workers (2021) detected SARS-CoV-2 neutralizing antibodies in a pangolin at a wildlife checkpoint in Southern Thailand. Hence, pangolins appear to be naturally infected by viruses from at least two sarbecovirus lineages, although their role, if any, in the genesis of SARS-CoV-2 is uncertain.
Investigations on the diversity of sarbecoviruses and other coronaviruses in bats and other species will provide critical data on the evolution and ecology of potential pathogens, guidance for detecting their emergence and suggest solutions for design of appropriate countermeasures. In this regard there have been suggestions that scientists should stop investigating the diversity of coronaviruses in bats and other animals (Baker, 2021). We contend that the world should do the opposite if we are to be better prepared to prevent the next pandemic of an emergent coronavirus.
Conclusions
Newly sequenced sarbecoviruses from bats captured in Cambodia, Thailand and Japan possess different combinations of spike motifs in the RBD and the S1/S2 junction that were first described in SARS-CoV-2. These observations are consistent with the natural origin of SARS-CoV-2 and strongly inconsistent with a laboratory origin. Studies of coronavirus diversity in bats and other species must continue.
References
Andersen KG, Rambaut A, Lipkin WI, Holmes EC, and Garry RF. (2020). The proximal origin of SARS-CoV-2. Nat Med 26, 450-452.
Baker N. (2021). The Lab-Leak Hypothesis. Was COVID-19 a Wuhan Lab Leak? A Coronavirus Investigation
Boni MF, Lemey P, Jiang X, Lam TT, Perry BW, Castoe TA, Rambaut A and Robertson DL. (2020). Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nature Microbiology 5, 1408-1417.
Chan YA and Zhan SH. (2020). Single source of pangolin CoVs with a near identical Spike RBD to SARS-CoV-2. https://www.biorxiv.org/content/10.1101/2020.07.07.184374v2.
Faria NR, Claro IM, Candido D, Moyses Franco LA, Andrade PS, Coletti TM, et al. (2021) Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: preliminary finding. https://pando.tools/t/586/2.
Garry RF. (2021). Mutations arising in SARS-CoV-2 spike on sustained human-to-human transmission and human-to-animal passage. https://pando.tools/t/578/5.
Garry RF, Andersen KG, Gallaher WR, Lam TT, Gangaparapu K, Latif AA, et al. (2021). Spike protein mutations in novel SARS-CoV-2 ‘variants of concern’ commonly occur in or near indels. https://pando.tools/t/605.
Ge X-Y, Wang N, Zhang W, Hu B, Li B, Zhang Y-Z, Zhou J-H, et al. (2016). Coexistence of multiple coronaviruses in several bat colonies in an abandoned mineshaft. Virologica Sinica 31, 31-40.
Hul V, Delaune D, Karlsson EA, Putita OT, Hassanin A, Baidaliuk A, et al. (2021). A novel SARS-CoV-2 related coronavirus sublineage in bats from Cambodia. https://doi.org/10.1101/2021.01.26.428212.
Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. (2020). Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215-220.
Lam TT, Jia N, Zhang YW, Shum MH, Jiang JF, et al. (2020). Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 583, 282-5.
Li F. (2008). Structural analysis of major species barriers between humans and palm civets for severe acute respiratory syndrome coronavirus infections. J Virol 82, 6984-91.
Li J-B, Liu H, Yin T-T, Peng M-S and Zhang Y-P. (2021). Unpublished observations.
Liu P, Chen W and Chen JP. (2019). Viral metagenomics revealed Sendai virus and coronavirus infection of Malayan pangolins (Manis javanica). Viruses 11, 979.
Murakami S., Kitamura T, Suzuki J., Sato R, Aoi T, Fujii M, et al. (2020). Detection and characterization of bat sarbecovirus phylogenetically related to SARS-CoV-2, Japan. Emerg Infect Dis 26, 3025-3029.
Piplai S, Singh PK, Winkler DA and Petrovsky N (2020). In silico comparison of spike protein-ACE2 binding affinities across species; significance for the possible origin of the SARS-CoV-2 virus. arXiv:2005.06199 [q-Bio.BM].
Rambaut A., Loman N, Pybus O, Barclay W, Barrett J, Carabelli A, Connor TR, Peacock T, Robertson DL, and Volz E. (2020). Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. https://pando.tools/t/563/5.
Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, et al. (2003). Characterization of a novel coronavirus associated with Severe Acute Respiratory Syndrome. Science 300, 1394-99.
Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, et al. (2020). Structural basis of receptor recognition by SARS-CoV-2. Nature 581, 221-224.
Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, et al. (2020). Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. https://doi.org/10.1101/2020.12.21.20248640 6.
Tian S. (2009). A 20 residues motif delineates the furin cleavage site and its physical properties may influence viral fusion. Biochemistry Insights 2, 9–20.
Wacharapluesadee S, Tan CW, Manee-Orn P, Duengkae P, Zhu F, Joyjinda Y, et al. (2021). SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nature Communications Nature Communications 12, 972.
Wu L, Chen Q, Liu K, Wang J, Han P, Zhang Y, et al. (2020). Broad host range of SARS-CoV-2 and the molecular basis for SARS-CoV-2 binding to cat ACE2. Cell Discovery 6, 68.
Wu Y and Zhao S. (2020). Furin cleavage sites naturally occur in coronaviruses. Stem Cell Res 50, 102115. doi: 10.1016/j.scr.2020.102115.
Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, et al. (2020). A new coronavirus associated with human respiratory disease in China. Nature 579, 265-269.
Xiao K, Zhai J, Feng Y, Zhou N, Zhang X, Zou JJ, et al. (2020). Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature 583, 286-289.
Zhan S, Deverman B, and Chan Y. (2020). SARS-CoV-2 is well adapted for humans. What does this mean for re-emergence? https://doi.org/10.1101/2020.05.01.073262.
Zhou H, Chen X, Hu T, Li J, Song H, Liu Y, et al. (2020). A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Curr Biol 30, 2196-2203.e2193.
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si H-R, et al. (2020a). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270-273.
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si H-R et al.. (2020b). Addendum: A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 588, E6 (2020). https://doi.org/10.1038/s41586-020-2951-z.