Spike protein mutations in novel SARS-CoV-2 ‘variants of concern’ commonly occur in or near indels.
Robert F. Garry1,2,*, Kristian G. Andersen3,4, William R. Gallaher5,6, Tommy Tsan-Yuk Lam7,8, Karthik Gangaparapu3,4, Alaa Abdel Latif3,4, Brandon J. Beddingfield9, Andrew Rambaut10 and Edward C. Holmes11
1.Department of Microbiology and Immunology, Tulane University Medical Center, 1430 Tulane Avenue, New Orleans, Louisiana 70112 USA.
2.Zalgen Labs, LLC, Germantown, MD, USA.
3.Department of Immunology and Microbiology, The Scripps Research Institute, La Jolla, CA 92037, USA.
4.Scripps Research Translational Institute, La Jolla, CA 92037, USA.
5.Mockingbird Nature Research Group, PO Box 568, Pearl River, LA 70452, USA.
6.Emeritus Faculty, Department of Microbiology, Immunology and Parasitology, Louisiana State University Health Sciences Center, 1901 Perdido Street, New Orleans, Louisiana 70112, USA
7.Joint Institute of Virology (Shantou University and The University of Hong Kong), Guangdong-Hongkong Joint Laboratory of Emerging Infectious Diseases, Shantou University, Shantou, P. R. China.
8.State Key Laboratory of Emerging Infectious Diseases, School of Public Health, The University of Hong Kong, Hong Kong, P. R. China.
9.Tulane School of Medicine, Tulane National Primate Research Center, 18703 Three Rivers Road, Covington, LA 70433, USA.
10.Institute of Evolutionary Biology, University of Edinburgh, Edinburgh, UK.
11.Marie Bashir Institute for Infectious Diseases and Biosecurity, School of Life and Environmental Sciences and School of Medical Sciences, The University of Sydney, Sydney, Australia.
*corresponding author: E-mail: [email protected].
Introduction
In recent months a number of genetically distinct variants of SARS-CoV-2, characterised by the accumulation of multiple amino acid replacements and insertion-deletion (indel) changes compared to their closest relatives, have appeared in different geographic locations. Because they have been associated with elevated rates of viral spread (Kupferschmidt, 2021), these lineages have also been termed ‘variants of concern’ (Volz et al., 2021; Naveca et al., 2021). SARS-CoV-2 lineage B.1.1.7 likely arose in the United Kingdom in September 2019 and is characterized by 17 mutations, including 8 in the spike protein (Rambaut et al., 2020). Other lineages, including B.1.351, initially detected in South Africa (Tegally et al., 2020), and most recently lineage P.1, first documented in the Amazonia region of Brazil (Faria et al., 2020), carry additional mutations. All three lineages are characterised by a N501Y mutation in the spike protein, while both B.1.351 and P.1 also carry the spike mutation E484K. In addition, both B.1.1.7 and B.1.351, but not P.1, have acquired short sequence deletions in the spike protein. Here, we examine the genomic location of lineage-defining mutations in the spike proteins of these newly emerged SARS-CoV-2 lineages and show that they generally occur in evolutionary ‘hotspots’.
Methods
The following representative sequences from the Sarbecovirus subgenus of the Betacoronaviruses (Coronaviridae), including SARS-related coronaviruses (SARSr CoV), were selected for comparative analysis: (i) Bat coronavirus RmYN02 (accession EPI_ISL_412977); (ii) Bat SARS-like coronavirus CoVZC45 (MG772933.1); (iii)Bat SARS-like coronavirus CoVZXC21 (AVP78042.1); (iv) Civet SARS-CoV 007/2004 (AAU04646.1); (v) SARS-CoV Urbani (AAP13441.1); (vi) Pangolin coronavirus GX-P1E (QIA48623.1); (vii) Vero E6 cell passaged Pangolin coronavirus GX-P2V (QIQ54048.1); (viii) Pangolin coronavirus GD-MP789 (QIG55945.1); (ix) Bat coronavirus RaTG13 (QHR63300.2); (x) SARS-CoV-2 South Africa/KRISP-MDSH920868/2020 (EPI_ISL_736967); (xi) SARS-CoV-2 England/LOND-1267020/2020 (EPI_ISL_741243); (xii) SARS-CoV-2 Brazil/Am-L70-CD1722/2020 (EPI_ISL_804832); and (xiii) SARS-CoV-2 Wuhan-Hu-1 (YP_009724390.1).
All spike amino acid sequences from these viruses were aligned using Clustal Omega (Sievers et al., 2011) and adjusted by visual inspection. The locations of the signal peptide cleavage sites were analyzed using SignalP v5.0 (Almagro Armenteros et al., 2019).
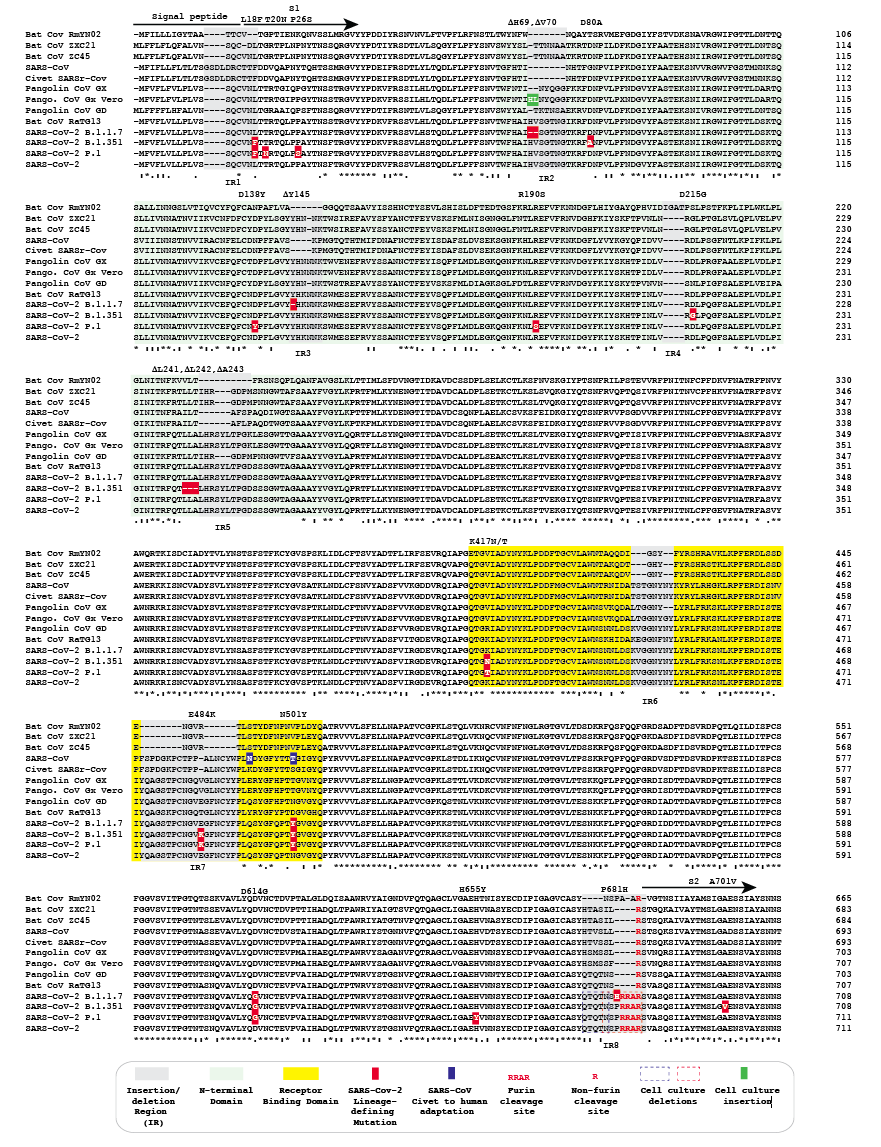
Figure 1. Amino acid alignment of spike protein sequences of sarbecoviruses. The signal peptide, S1 subunit and part of S2 are shown. The remainder of the alignment in S2 does not have additional insertions or deletions, but does contain the B.1.1.7 substitutions T716I, S982A, D1118H and P.1 substitution T1027I.
Results
Alignment of the amino acid sequences of representative sarbecovirus spike proteins identified several regions that have acquired indels (Fig. 1). While caution should be exercised in ascribing evolutionary pathways to the presence or absence of any indel, it is apparent that there are several locations that are seemingly prone to the gain or loss of short nucleotide sequences as sarbecoviruses have been transmitted among animals and humans from their likely progenitor viruses in bats. Relative to other CoVs, bat SARSr CoV RmYN02 generally contains larger deletions than other bat CoVs. However, RmYN02 spike also includes a 4 amino acid sequence that is absent from other bat CoVs as well as known animal and human sarbecoviruses. For the purpose of this analysis the RmYN02 spike sequence was used to define the boundaries of indel regions in sarbecoviruses (grey highlights, Fig. 1). Indels from all the other sarbecoviruses analyzed fall either within these boundaries or immediately adjacent to these indel regions.
SARS-CoV, as well as SARSr CoVs from pangolins, displays a distinct subset of spike indels compared to bat CoVs or SARS-CoV-2. SARS-CoV is a direct zoonosis from a closely related virus in civets and horseshoe bats (Li, 2008). The spike protein sequence of SARSr-CoV of civets does not have any indels relative to the SARS-CoV spike. Sarbecoviruses isolated from pangolins share common ancestry with the bat sarbecovirus RaTG13 and SARS-CoV-2 (Liu et al., 2019; Lam et al., 2020; Xiao et al., 2020; Boni et al., 2020). Two lineages of sarbecoviruses, designated here as GX (Guangxi) and GD (Guangdong) have been isolated from illegally imported Malayan pangolins, Manis javanica, which are non-native to China. The GX and GD pangolin CoV spike proteins analysed differ in the length of sequence in 4/8 indel regions. With the exception of the insertion that generated the furin cleavage site, all of the sequences in the Bat CoV RaTG13 spike corresponding to indel regions are the same length as the corresponding sequences in SARS-CoV-2 spike.
Inspection of the alignment of sarbecovirus spikes reveals that many lineage-defining mutations in the newly emerged SARS-CoV-2 variants of concern occur in or adjacent to these indels. Specifically:
(a) Indel region 1 is in a variable sequence that overlaps the signal peptide and the beginning of the S1 spike subunit. The spike protein sequences of SARS-CoV and the SARSr CoV of civets contain a 4 amino acid putative insertion in this region. Compared to the RmYN02 spike protein, each of the other bat, human, civet and pangolin CoVs contain one or two amino acid insertions following a conserved cysteine predicted to be the terminal amino acid of the spike signal peptide. L18F in lineages B.1.351 and P.1 is in this indel region. P.1 also carries T20N and P26S, which are located in the variable region that includes indel region 1.
(b) Indel region 2 occurs near the beginning of the N-terminal domain (NTD). Relative to RmYN02, all other spike proteins analyzed, have insertions of variable lengths (5, 6 or 7 amino acids) in this indel region with the exception of SARS-CoV and the SARSr CoV from civets. Lineage B1.1.7 deletions delH69 and delV70 occur in this indel region and the D80A substitution in lineage B.1.351 lies downstream. Vero cell passage of a GX pangolin CoV resulted in a two amino acid insertion relative to other GX spikes at the location corresponding to B1.1.7 delH69 and delV70 (Fig. 1 green highlight).
(c) Indel region 3 is located in the central part of the NTD. Relative to the RmYN02 spike protein, all other sarbecovirus spike protein sequences analyzed have insertions in this region (2, 5 or 6 amino acids). B1.1.7 delY145 occurs in this indel region, while the D138Y substitution in lineage P.1 is upstream.
(d) Indel region 4 represents the only putative insertion (4 amino acids) found in RmYN02 relative to the other spike protein sequences analyzed. The D215G mutation in B.1.351 is adjacent to this indel region.
(e) Indel region 5 is located near the end of the NTD. Relative to RmYN02, all the other spike protein sequences analyzed have insertions in this region (4, 8 or 11 amino acids). The B.1.351 isolate depicted has delL241, delL242 and delA243 adjacent to this indel region. Other B.1.351 isolates from South Africa carry L242H and R246I mutations that are also near or in this indel region (Tegally et al., 2020).
(f) Indel region 6 is located in the receptor binding domain (RBD). While no mutations in the SARS-CoV-2 lineages with multiple spike mutations occur in this region, a conserved tyrosine residue adjacent to this indel region has been noted to change on occasion with human-to-mink or human-to-domestic cat transfer (Y453F).
(g) Indel region 7 is located in the RBD. Relative to the Bat CoV RmYN02, ZXC21 and ZC45 spike proteins, the spike proteins of SARS-CoV and the SARSr CoV of civets have a 15 amino acid insertion and Bat CoV RaTG13 and other analyzed sarbecovirus spikes have a 16 amino acid insertion. Both lineages B.1.351 and P.1 carry a E484K amino acid substitution that is located in this indel region.
(h) Indel region 8, located at the S1/S2 junction, has been the subject of considerable discussion (Andersen et al., 2020; Coutard et al., 2020; Gallaher, 2020a). Alignment of the spike protein of SARS-CoV-2 with those of RaTG13 and other sarbecoviruses demonstrates that a 12 base insertion in the SARS-CoV-2 spike gene adds 4 amino acids (PRRA) at the S1/S2 junction and converts a monobasic cleavage site [R] to a minimal furin (polybasic) cleavage site (RRAR). The P681H amino acid substitution in lineage B.1.1.7 is located in this indel region. Passage of SARS-CoV-2 in Vero cells can result in deletion of either the furin cleavage site or a sequence immediately adjacent to it (Liu et al., 2020, Lau et al., 2020).
Amino acid substitutions R190S, K417N/T, N501Y, D614G, H655Y and A701V occur in SARS-CoV-2 lineages B.1.1.7, B.1.351 and P.1, but lie outside of indel regions. D614G and N501Y are notable as the only mutations common to each of the three lineages. D614G variants appeared prior to the detection of the multi-mutation variants of concern, are thus not specific for these lineages, and likely provide a fitness advantage (Plante et al, 2020). N501Y appears to provide not only a fitness advantage, but also may contribute to immune escape (Starr et al., 2010; Greaney et al., 2012). N501 is also the SARS-CoV2 homolog to one of two residues K479N and S487T (Fig. 1, blue highlights) that are mutated during adaptation of the sarbecovirus of civets to humans (Li, 2008).
Discussion
Short insertions or deletions have impacted evolution of CoV spike during interspecies transfers. These indels remain hotspots for mutational events, as shown by the emergence of several variants of concern that bring several mutations together in various constellations that can impact transmissibility and/or lead to immune escape (Wibmer et al., 2021; Wang et al., 2021). Most of these indel regions occur at or adjacent to exterior peptide loops that are able to accept changes without disturbing the framework structure of the S1 protein domains. However, not all such loop peptide regions are affected. The 8 indel regions appear to preferentially affect external loops that may be in contact with ligands to S1, and with the furin protease (Gallaher, 2020a, Qing et al., 2020). The potential role of variable loops in immune escape and viral fitness have been the subject of a large body of research on other viruses, including HIV and influenza virus. In this regard, amino acid substitutions in the spike proteins in the recently emerged variants of concern often involve purine/pyrimidine transversions and result in nonconservative amino acid changes suggesting that they arose under selective pressure.
While the insertion that generated the furin cleavage site in the spike has been a strong focus of attention due to its impact on SARS-CoV-2 fitness and transmissibility (Johnson et al., 2020; Lau et al., 2020; Chu et al., 2021), sarbecovirus evolution has involved several additional insertions and deletions of short sequences in spike. Analysis of multi-mutation variants that have arisen reveals that regions containing these indels remain evolutionarily active as SARS-CoV-2 is spreading through the human population. Mutations in or near indel regions 1-8 in multi-mutant variants B.1.1.7, B.1.351 and P.1 have arisen during extended human-to-human transmission. Changes in or near these variable regions have also occurred during interspecies transfers of SARS-CoV and during transfers of SARS-CoV-2 from humans to mink and other species (Garry, 2021).
Comparison of the spike protein sequence of RmYN02 with those of other sarbecoviruses demonstrates that indel region 8 at the S1/S2 junction is highly variable (Zhou et al., 2020). Insertion or deletion events in or near the furin cleavage site are a frequent occurrence during coronavirus evolution (Garry and Gallaher, 2020). Hence, analyses suggesting that the evolutionary origins of the RmYN02 S1/S2 cleavage site can be revealed by a simple nucleotide alignment (Segreto and Deigin, 2020) are overly simplistic. The current analysis shows that indel regions 1, 6 and 7, like indel region 8, are complex. These four evolutionarily volatile regions have been subjected to more than one and possibly several insertion or deletion events during sarbecovirus evolution, which cannot be defined by superficial analyses of the underlying nucleotide sequence.
Some proponents of a laboratory origin of SARS-CoV-2 have argued that the insertion in indel region 8 could have been generated by passage in cell culture. However, the only types of in-frame modifications to indel region 8 that have thus far been reported to occur after cell culture passage are deletions. These include a deletion of the furin cleavage site itself (NSPRRAR) and a deletion of an upstream sequence (QTQTN) that may be involved in recognition of the cleavage site (Liu et al., 2020; Lau et al., 2020). Indel 8 in SARS-CoV-2 appears to remain under evolutionary pressure during human-to-human transmission. The lineage B.1.1.7 P681H amino acid substitution in this region replaces the non-optimal P at the furin cleavage site with a more favorable positively charged H (Tian and Jianhua, 2010). Independent variants carrying the P681H substitution have been detected in Nigeria and elsewhere (Happi et al., 2020).
Insertions have been generated in the SARS-CoV-2 spike protein after passage in Vero cells, but these occur near indel region 4 rather than indel region 8. This is the only SARS-CoV-2 spike indel showing a deletion relative to the RmYN02 spike protein. An insertion of 4 amino acids - KLRS - following D215 is the dominant variant that arose after 6 passages of SARS-CoV-2 in Vero cells (Gangaparapu et al., unpublished observations). Similarly, hCoV-19/Slovenia/751/2020 (EPI_ISL_635200|2020-03-05), which was also passaged on Vero cells, has a AKKN insertion before D215. While short deletions have occurred in lineages B.1.1.7 and B1.351 in or near indel regions 2, 3 and 5 and are not uncommon in other lineages, insertions in SARS-CoV-2 spike are relatively rare overall. Of over 350,000 SARS-CoV-2 sequences in the GSAID database, only 51 list insertions in the spike protein. Most of these appear to be sequencing anomalies. However, two hCoV-19/England/MILK-B8C845/2020 (EPI_ISL_675778) and hCoV-19/England/ALDP-CB0759/2020 (EPI_ISL_760951) have an insertion at R214 adjacent to indel 4 indicating that this region of SARS-CoV-2 spike can accommodate short insertions.
Pangolins captured in the wild carry at least two related, but distinct, sarbecovirus lineages (Liu et al., 2019; Lam et al., 2020; Xiao et al., 2020; Boni et al., 2020). The spike proteins of the GX and GD lineages display a disparate indel structure. Recently, an additional sarbecovirus has been sampled from a Chinese pangolin, Manis pentadactyla, collected in 2017 in Yunnan province, China (GISAID ID EPI_ISL_610156). The spike gene sequence was incompletely sequenced in the CoV detected in this animal, precluding comparisons. Further studies of CoVs from pangolins should be a priority to further elucidate the evolutionary and ecological relationships of these viruses with CoVs of bats and other animals. Moreover, the GX lineage of pangolin CoVs provides another example of insertion following passage of a sarbecovirus in Vero cells. Pangolin CoVs of the GX lineage inserted two amino acids in indel region 2 following passage in Vero E6 cells (Lam et al., 2020, Fig. 1). This insertion occurs at the same location as delH69 and delV70 in lineage B.1.1.7, which has been suggested to provide a fitness advantage during human-to-human passage of SARS-CoV-2 (Kemp et al., 2020). The H69-V70 deletion has also been detected in the cluster-5 variant that arose after transfer of SARS-CoV-2 from humans to commercially raised mink (Koopmans, 2021).
Conclusions
Although CoVs have a proof-reading apparatus (Robson et al., 2020), their genomes remain subject to recombination as well as other copy-choice transcriptional errors (Gallaher, 2020b). It will be important to elucidate genomic features that favor the formation of short insertions or deletions in CoV genomes and to define molecular processes occurring during CoV genome replication that produce indels. Analyses of natural sarbecoviruses in bats and other species will be essential to addressing these questions. Such studies can potentially provide further insight into the evolutionary pathways that generate new CoV variants, and. recent analyses suggest the existence of as yet unsampled sarbecovirus lineages (Boni et al., 2020). As discussed by Lytras et al. (2021), we contend that calls for a moratorium on identification of new CoVs in bats and other wild animals (for example: Baker, 2021) are ill-considered.
Acknowledgments
We thank all those who have contributed genome sequences to the GISAID database (https://www.gisaid.org/) and analyses and ideas to Virological.org (http://virological.org/). RFG is supported by the National Institutes of Health (U19AI135995, U54 HG007480 and U19AI142790), the Coalition for Epidemic Preparedness Innovations, the Burroughs Wellcome Fund, the Wellcome Trust, the Center for Disease Prevention and Control, and the European & Developing Countries Clinical Trials Partnership.KGA is a Pew Biomedical Scholar and is supported by NIH NIAID grant U19AI135995. TTYL is supported by Excellent Young Scientists Fund (Hong Kong and Macau) (31922087) from The Natural Science Foundation of China. AR is supported by the Wellcome Trust (Collaborators Award 206298/Z/17/Z – ARTIC network) and the European Research Council (grant agreement no. 725422 – ReservoirDOCS). ECH is supported by an ARC Australian Laureate Fellowship (FL170100022).
References
Almagro Armenteros JJ, Tsirigos KD, Sonderby CK, Petersen TN, Winther O, Brunak S, et al. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol. 37, 420-3.
Andersen KG, Rambaut A, Lipkin WI, Holmes EC, and Garry RF. (2020). The proximal origin of SARS-CoV-2. Nat Med 26, 450-452.
Baker N. (2021). The Lab-Leak Hypothesis. Was COVID-19 a Wuhan Lab Leak? A Coronavirus Investigation.
Boni MF, Lemey P, Jiang X, Lam TT, Perry BW, Castoe TA, Rambaut A and Robertson DL. (2020). Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nature Microbiology 5, 1408-1417.
Chu H, Hu B, Huang X, Chai Y, Zhou D, Wang Y, et al. (2021). Host and viral determinants for efficient SARS-CoV-2 infection of the human lung. Nature Communications. 12, 134.
Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, and Decroly E. (2020). The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res 176, 104742.
Faria NR, Claro, IM, Candido D, Moyses Franco LA, et al. (2020). Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: preliminary findings. https://pando.tools/t/genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-manaus-preliminary-findings/586.
Gallaher WR. (2020a). Similarities between the SARS-CoV-2 spike protein lima bean lection specific for ABO blood group A. https://pando.tools/t/similarities-between-sars-cov-2-spike-protein-and-lima-bean-lectin-specific-for-abo-blood-group-a/518.
Gallaher WR (2020b). A palindromic RNA sequence as a common breakpoint contributor to copy-choice recombination in SARS-COV-2. Arch Virol 165, 2341-8.
Garry RF. (2021). Mutations arising in SARS-CoV-2 spike on sustained human-to-human transmission and human-to-animal passage. https://pando.tools/t/mutations-arising-in-sars-cov-2-spike-on-sustained-human-to-human-transmission-and-human-to-animal-passage/578/5.
Garry RF. and Gallaher WR (2020). Naturally occurring indels in multiple coronavirus spikes. https://pando.tools/t/naturally-occurring-indels-in-multiple-coronavirus-spikes/560.
Greaney AJ, Starr TN, Gilchuk P, Zost SJ, Binshtein E, Loes AN, et al. (2021). Complete mapping of mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that escape antibody recognition. Cell Host & Microbe, 29, 44-57.e9.
Happi C, Ihekweazu ., Nkengasong J, Oluniyi PE, and Olawoye I. (2020). Detection of SARS-CoV-2 P681H Spike Protein Variant in Nigeria. Detection of SARS-CoV-2 P681H Spike Protein Variant in Nigeria. https://pando.tools/t/detection-of-sars-cov-2-p681h-spike-protein-variant-in-nigeria/567
Johnson BA, Xie X, Kalveram B, Lokugamage KG, Muruato A, Zou J, et al. (2020). Furin cleavage site Is key to SARS-CoV-2 pathogenesis. doi.org/10.1101/2020.08.26.268854.
Kemp, S.A., Harvey, W.T., Datir, R.P., Collier, D.A., Ferreira, I., Carabelli, A.M., Robertson, D.L., and Gupta, R.K. (2020). Recurrent emergence and transmission of a SARS-CoV-2 Spike deletion ΔH69/V70. bioRxiv 2020.12.14.422555; doi: https://doi.org/10.1101/2020.12.14.422555
Koopmans, M. (2021). SARS-CoV-2 and the human-animal interface: outbreaks on mink farms. The Lancet Infectious Diseases 21, 18-19.
Kupferschmidt K. (2021). New coronavirus variants could cause more reinfections, require updated vaccines
doi:10.1126/science.abg6028. Online before print Jan. 15, 2021.
Lam TT, Jia N, Zhang YW, Shum MH, Jiang JF, Zhu HC, et al. (2020). Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 583, 282-5.
Lau SY, Wang P, Mo, BW, Zhang AJ, Chu H, Lee AC, Deng S, Chen P, Chan KH, Song W, et al. (2020). Attenuated SARS-CoV-2 variants with deletions at the S1/S2 junction. Emerging Microbes & Infections 9, 837-842.
Li F. (2008). Structural analysis of major species barriers between humans and palm civets for severe acute respiratory syndrome coronavirus infections. J Virol 82, 6984-91.
Liu P, Chen W and Chen, JP. (2019). Viral metagenomics revealed sendai virus and coronavirus infection of Malayan pangolins (Manis javanica). Viruses 11, 979.
Liu Z, Zheng H, Lin H, Li M, Yuan R, Peng J, Xiong Q, Sun J, Li B, Wu J, et al. (2020). Identification of common deletions in the spike protein of severe acute respiratory syndrome coronavirus 2. J Virol 94.:e00790-20. doi: 10.1128/JVI.00790-20.
Lytras S, Hughes J, Jiang X, and Robertson DL. (2021). Exploring the natural origins of SARS-CoV-2. https://pando.tools/t/exploring-the-natural-origins-of-sars-cov-2/595.
Naveca F, da Costa C, Nascimento V, Souza V, Corado V, Nascimento F, Costa A, et al. (2021). SARS-CoV-2 reinfection by the new Variant of Concern (VOC) P.1 in Amazonas, Brazil. https://pando.tools/t/sars-cov-2-reinfection-by-the-new-variant-of-concern-voc-p-1-in-amazonas-brazil/596.
Plante JA, Liu Y, Liu J, Xia H, Johnson BA, Lokugamage KG, Zhang X, Muruato AE, Zou J, Fontes-Garfias CR, et al. (2020). Spike mutation D614G alters SARS-CoV-2 fitness. Nature. epublication 10.1038/s41586-020-2895-3.
Qing E, Hantak M, Perlman S, and Gallagher T. (2020). Distinct roles for sialoside and protein receptors in coronavirus infection. mBio. 2020 Feb 11;11(1):e02764-19. doi: 10.1128/mBio.02764-19.
Rambaut A., Loman N, Pybus O, Barclay W, Barrett J, Carabelli A, Connor TR, Peacock T, Robertson DL, and Volz E. (2020). Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. https://pando.tools/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563/5.
Robson F, Khan Ks, Le TK, Paris C, Demirbag S, Barfuss P et al. (2020). Coronavirus RNA proofreading: Molecular basis and therapeutic targeting. Molecular Cell 79, 710-27.
Segreto R and Deigin Y. (2020). The genetic structure of SARS‐CoV‐2 does not rule out a laboratory origin: SARS‐COV‐2 chimeric structure and furin cleavage site might be the result of genetic manipulation. BioEssays. DOI:10.1002/bies.202000240
Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KHD, Dingens AS, et al. (2020). Deep mutational scanning of SARS-CoV-2 Receptor Binding Domain reveals constraints on folding and ACE2 binding. Cell 182, 1295-310.e20.
Tian S and Jianhua W. (2010). Comparative study of the binding pockets of mammalian proprotein convertases and its implications for the design of specific small molecule inhibitors. International Journal of Biological Sciences 6, 89-95.
Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, et al. (2020). Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. https://doi.org/10.1101/2020.12.21.20248640.
Volz E, Mishra S, Chand M, et al. (2021). Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. bioRxiv. 2021; published online Jan 4. DOI:10.1101/2020.12.30.20249034.
Wang, Z., Schmidt, F., Weisblum, Y., Muecksch, F., Barnes, C.O., Finkin, S., Schaefer-Babajew, D., Cipolla, M., Gaebler, C., Lieberman, J.A., et al. (2021). mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. https://doi.org/10.1101/2021.01.15.426911.
Wibmer, C.K., Ayres, F., Hermanus, T., Madzivhandila, M., Kgagudi, P., Lambson, B.E., Vermeulen, M., van den Berg, K., Rossouw, T., Boswell, M., et al. (2021). SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. https://doi.org/10.1101/2021.01.18.427166.
Xiao K, Zhai J, Feng Y, Zhou N, Zhang X, Zou JJ, et al. (2020). Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature 583, 286-9.