Sandra Martínez-Puchol 1,2,3 , Andreu Coello 2,3 , Antoni E. Bordoy 2,3 , Laia Soler 3 , David Panisello 2 , Sara González-Gómez 2 , Gemma Clarà 3 , Alexia Paris de León 3 , Anna Not 2 , Águeda Hernández 3 , Silvia Bofill-Mas 4,5 , Verónica Saludes 2,3,6,* , Elisa Martró 2-4,6,* , Pere Joan Cardona 2,3,7,*
- Vicerectorat de recerca, Universitat de Barcelona, Universitat de Barcelona (UB).
- Fundació Institut d’Investigació en Ciències de la Salut Germans Trias i Pujol (IGTP), Badalona, Spain.
- Microbiology Department, Northern Metropolitan Clinical Laboratory, Hospital Universitari Germans Trias i Pujol (HUGTiP), Badalona, Spain.
- Laboratory of Virus Contaminants of Water and Food, Section of Microbiology, Virology and Biotechnology, Department of Genetics, Microbiology and Statistics, Faculty of Biology,University of Barcelona, Spain.
- The Water Institute of the University of Barcelona, Spain
- CIBER in Epidemiology and Public Health (CIBERESP), Madrid, Spain.
- CIBER in Respiratory Diseases (CIBERES), Madrid, Spain.
*Correspondence to: [email protected], [email protected], [email protected]
Although Monkeypox virus (MPXV) is usually constrained to small outbreaks in remote parts of Central and West African countries, in May 2022 multiple cases of MPXV in several non-endemic countries have been shown to be phylogenetically related.
As of 27th May 2022, eighty-four confirmed MPXV cases have been reported in Spain (https://monkeypox.healthmap.org/). The Spanish case reported here corresponds to a young individual fulfilling clinical and epidemiological criteria. Whole-genome sequencing and phylogenetic analysis were performed. A first draft genome with 98.6% coverage and an average depth of 77x was reconstructed. Preliminary phylogenetic analysis of this genome together with MPVX genomes publicly available from Portugal, Belgium, France, Germany and the Netherlands showed evidence of a close genetic relationship within the West African clade. Specifically, ≤4 SNPs were identified versus Portugal sequences, and two SNPs versus the German sequence (Fig. 1).
Methods
Briefly, the clinical sample (skin swab in 400 l of viral transport media) was frozen and thawed three times to disrupt cell membranes. After a centrifugation step at 3000 rpm for 5 min, supernatant was treated with DNAse and RNAse [1]. Nucleic acid extraction was performed using QiAmp Viral RNA Mini Kit (Qiagen®) from DNase/RNase-treated clinical material, eluting in 80 μL AVE-buffer. Sequence-independent single primer amplification (SISPA) was performed on 32 μL of the extracted DNA [1]. Then, the DNA sequencing library was prepared from the SISPA purified product (85.6 ng of DNA) using the Ligation Sequencing Kit (SQK-LSK109, NEB®) with a final volume of 15 μL and a DNA quantity of 14.5 ng. The library was loaded onto an R9.4.1 flow cell which run for 19 hours on a MinION Mk1C device.
Human reads were excluded from the analysis using kraken2. The remaining reads were mapped against a reference genome (MT903343) using minimap2. Reads were sorted and indexed using samtools. A consensus was generated using iVar with a 10x coverage cut-off. Consensus sequences were aligned using Mafft and a SNP phylogenetic tree was build using IQ-TREE using the HKY+F model as previously described for ease of comparison (https://pando.tools/t/first-french-draft-genome-sequence-of-monkeypox-virus-may-2022/819). The sequence has been submitted to NCBI but is also provided here for early access.
MPXV_ES0001_HUGTiP_2022.zip (61.6 KB)
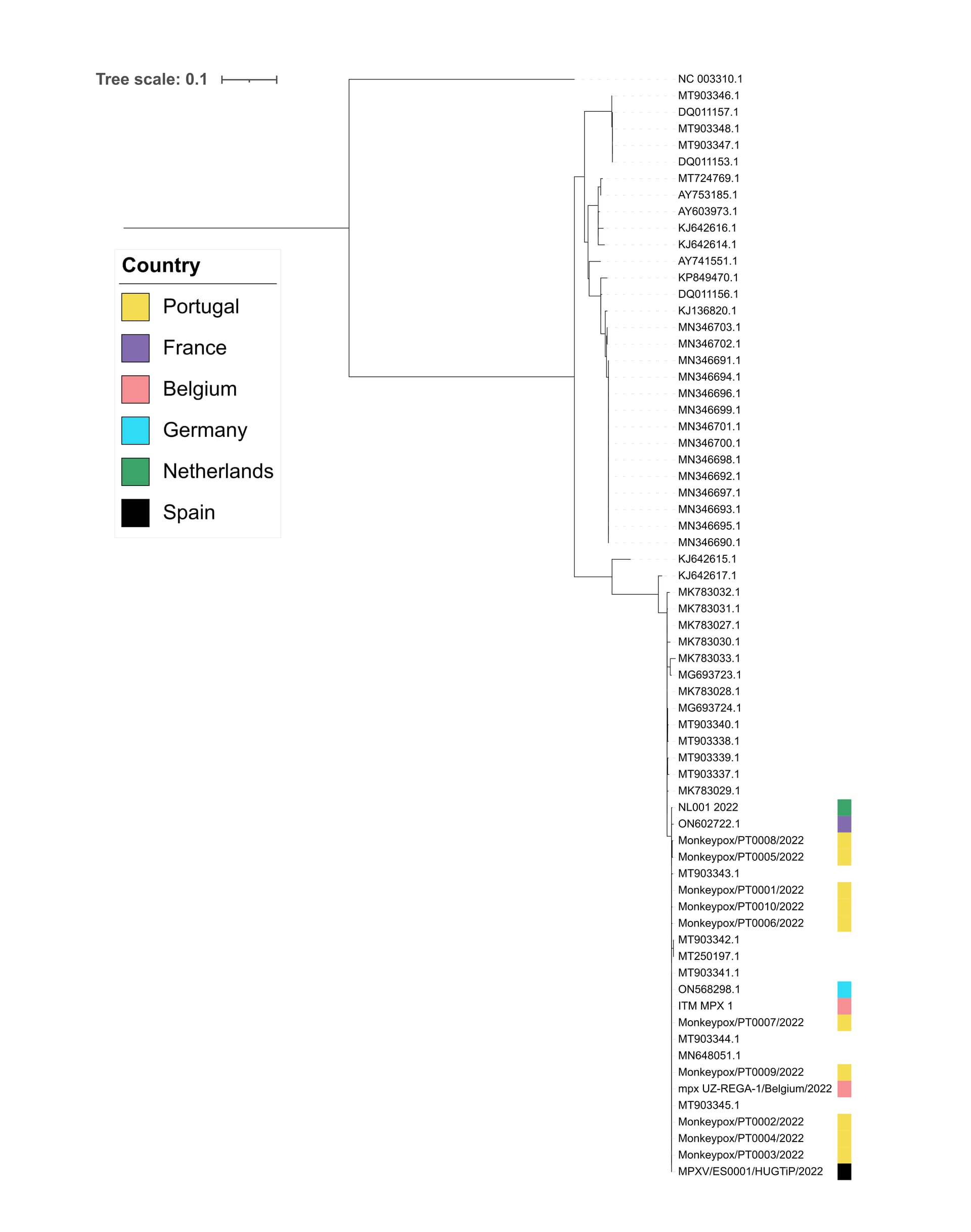
Figure 1. Draft maximum-likelihood phylogenetic tree obtained from the SNP alignment.
References
- Fernández-Cassi X., Rusiñol M., Martínez-Puchol S. Ultracentrifugation, and random amplification procedure to study the human blood virome. Part of the Methods in Molecular Biology book series (MIMB, volume 1838). The Human Virome: Methods and Protocols. Springer 2018