UPDATE: Two draft genomes from Madrid, Spain, of the Monkeypox virus 2022 outbreak
Sergio Buenestado-Serrano1, Rosalía Palomino-Cabrera1, Daniel Peñas-Utrilla1, Marta Herranz-Martín1, Alejandro Cobos1, Cristina Ventimilla1, Pilar Catalán1, Roberto Alonso1,2, Patricia Muñoz1,2, Laura Pérez-Lago1, Darío García de Viedma1,2
1 Clinical Microbiology and Infectious Diseases Department, Gregorio Marañón University Hospital, Gregorio Marañón Biomedical Research Institute, Madrid, Spain
2 CIBER Enfermedades Infecciosas (CIBERES), Spain
Monkeypox virus (MPXV) is a zoonotic virus endemic to West and Central Africa, which causes symptoms similar to those of smallpox, a disease that has been eradicated. At the beginning of May 2022, several countries began to report MPXV cases. Consequently, different laboratories started to sequence positive samples in order to study their phylogenetic relationship. As of 7th June 2022, 129 virus genome sequences have already been deposited at GISAID. It has allowed to identify 46 SNPs shared by all of them and not found in the genomes from the previous 2018/2019 outbreak (WHO report, Multi-country monkeypox outbreak in non-endemic countries 9).
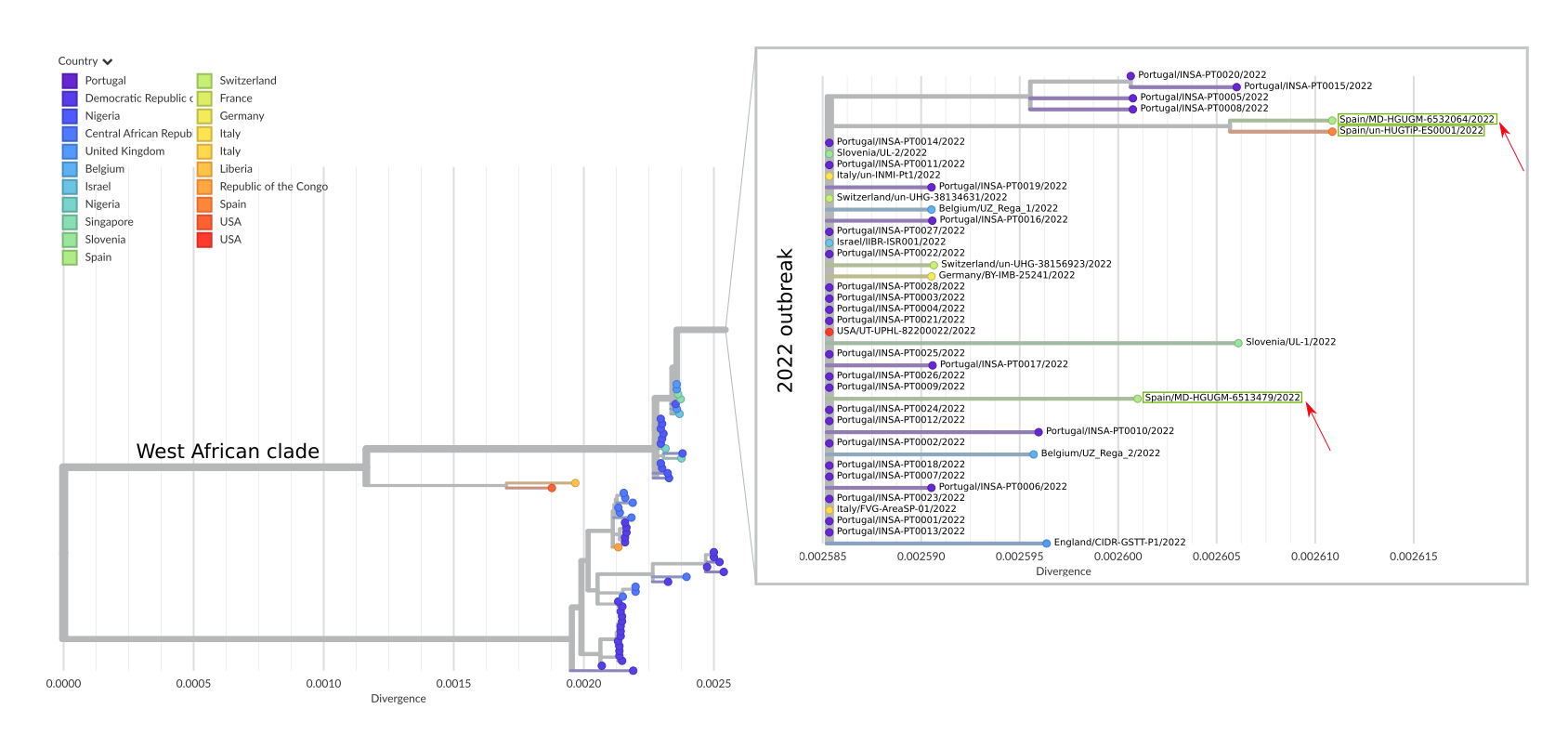
Figure 1. Draft phylogenetic tree of all 129 available Monkeypox genomes, including the 46 sequences from the 2022 outbreak and the previous ones. The zoomed window corresponds to the current outbreak; the three sequences from Spain (two from Madrid and one from Barcelona) are highlighted within green boxes and the sequences from Madrid (hMpxV/Spain/MD-HGUGM-6513479/2022 and hMpxV/Spain/MD-HGUGM-6532064/2022) are marked with red arrows. The tree was constructed with the Nexstrain monkeypox pipeline (GitHub - nextstrain/monkeypox: Nextstrain build for monkeypox virus 7).
The first draft genome reported (hMpxV/Spain/MD-HGUGM-6513479/2022) corresponds to a 36 year-old male, HIV-negative, with a non-severe clinical evolution. The average coverage was 15X, 61% of the genome was covered >10X and 95% was covered >5X. Of the 46 positions specific to the 2022 outbreak (Isidro, J. & al. 2022 1), 44 were identified with our pipeline. The raw fastq sequence has been submitted to ENA, and it can be found under the Run accession: ERR9814353 and the fasta has been uploaded to GISAID under the accession ID EPI_ISL_13089461.
This second draft genome (hMpxV/Spain/MD-HGUGM-6532064/2022) corresponds to a 31 year-old male, HIV-positive, with non-severe clinical evolution. For this second analysis, pre-sequencing laboratory procedures were optimized, which led to improving markedly sequencing depth values. The average coverage was 237X, with a depth > 30X for 100% of the genome. The raw fastq sequence has been uploaded to ENA under the Run accession ERR9821256. The fasta sequence was submitted to GISAID with accession ID EPI_ISL_13106454. This draft genome contains all 46 positions described as specific for the 2022 outbreak (Isidro, J. & al. 2022 1), in addition to 5 private SNPs. 6 SNPs were identified between the two draft genomes from Madrid.
Preliminary phylogenetic analysis (Figure 1) from our sequenced specimens shows that the obtained genomes belong to the West African clade of MPXV and are most closely related to the uploaded genomes from the 2022 outbreak. Both genome sequences were mapped against the other Spanish sequence published (ON622718.1); 6 and 2 SNPs were identified between the first and second Madrid drafts and the draft from Barcelona, respectively, indicating microevolution within this outbreak.
Methods
Briefly, a swab from a skin lesion was introduced in 1ml of viral transport medium. It was sonicated in a bath (two cycles of 20 seconds) and centrifuged at 3000 rpm for 5 minutes. The supernatant was treated with a DNase I/RNase A mixture for 40 min at 37°C and inactivated at 65°C/15 min and 1 min on ice. DNA was extracted using the automatic EMAG (Biomerieux) extractor and eluted in 100 µl. The sample was concentrated with AMPure XP beads (1:1) and eluted in 12 µl of water. A library was prepared using the Rapid Barcoding kit (SQK-RBK004; Nanopore), 50.6 ng of the library were loaded in a R9.4.1 flow cell and run on a MinION Mk1B device for 28 h.
Basecalling was performed using guppy v6.1.7, then a quality control of the sequences was performed with NanoFilt v2.8.0. Human reads (78.47% of the total reads) were depleted with Kraken2 v2.1.2. The remaining reads were mapped against the Monkeypox virus reference genome (MPXV_USA_2022_MA001; ON563414.3) using minimap2 v2.24. Subsequently, variant calling and consensus creation were made with samtools v1.15 and ivar 1.3.1.