The African Centre of Excellence for the Genomics of Infectious Disease [ACEGID], Redeemer’s University, Ede, Nigeria [RUN], and the Nigeria Centre for Disease Control [NCDC] report twenty [20] additional genome sequences of SARS-CoV-2 from Nigeria. These sequences are available at: [GitHub - acegid/CoV_Sequences: SARS-CoV-2 genome from Nigeria].
As a member of the Molecular Laboratory Network of the Nigeria Centre for Disease Control [NCDC], clinical specimens [specifically saliva, nasopharyngeal and nasal swabs] from suspected COVID-19 cases were sent to ACEGID, Redeemer’s University, for confirmatory testing, sequencing and molecular characterization. Viral RNA was extracted using the QiAmp viral RNA mini kit [Qiagen]. RT-qPCR was carried out at ACEGID using the DAAN RT-qPCR assay which confirmed the presence of SARS-CoV-2 viral RNA. Metagenomic sequencing libraries were prepared from total RNA as we previously described [Matranga et al ., 2016], and sequenced using the two Illumina MiSeqs in the sequencing platform of ACEGID.
Genome Assembly and Quality control
We carried out genome assembly using our publicly available software [viral-ngs v2.0] implemented on the DNA nexus cloud-based platform. We assembled 20 genomes [17 full and 3 partials]. We carried out quality control using fastqc (Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data).
Phylogenetics and Lineage Delineation
All HCoV whole genome sequences obtained from human hosts with geographical annotations were obtained from GISAID and aligned with the 17 full genomes from Nigeria.
Due to the large size of our tree, we couldn’t add the tree to this post, instead we constructed a smaller tree from an alignment of the new Nigerian sequences and global sequences that they clustered closely with. The smaller tree still reveals the same relationships as in the larger tree [Fig. 1].
Using Pangolin software [Rambaut et al ., 2020], we assigned the sequences to global SARS-CoV-2 lineages, and this revealed three different lineages [A, B.1 and B.2.1] of the virus circulating in Nigeria [Table 1]. Sequences from these lineages from Nigeria are clustering with sequences from Asia, Europe, USA and other African countries [Fig. 1], indicating multiple introduction of multiple lineages [Table 1] of the virus into the country.
Few of our sequences clustered together and formed a separate clade which strongly suggests local community transmission [Fig. 1]. Epidemiological data confirmed that all these sequences are from patients who travelled together in a community in Osun State, Nigeria. These findings further emphasize the power of genomics in elucidating community transmission during pandemics.

Figure 1 : Maximum likelihood tree of SARS-CoV-2. These sequences were aligned using MAFFT v7.310 [Katoh et al., 2009] and tree reconstruction using FastTree v2.1.11 [Price et al ., 2009]. The tip shapes (circle) representing the new Nigerian sequences are coloured green.
SARS_CoV_2_subset_aligned_tree.pdf (144.8 KB)
Table 1 : SARS-CoV-2 Lineages Circulating in Nigeria
| S/N | Taxon | Lineage | SH-alrt | UFbootstrap |
|---|---|---|---|---|
| 1. | Nigeria (CV35) | B.2.1 | 100 | 97 |
| 2. | Nigeria (CV34) | B.2.1 | 100 | 100 |
| 3. | Nigeria (CV29) | B.1 | 100 | 100 |
| 4. | Nigeria (CV24) | B.1 | 100 | 100 |
| 5. | Nigeria (CV22) | B.2.1 | 100 | 98 |
| 6. | Nigeria (CV21) | A | 100 | 100 |
| 7. | Nigeria (CV20) | A | 100 | 100 |
| 8. | Nigeria (CV18) | A | 100 | 100 |
| 9. | Nigeria (CV17) | A | 100 | 100 |
| 10. | Nigeria (CV14) | A | 100 | 100 |
| 11. | Nigeria (CV12) | A | 100 | 100 |
| 12. | Nigeria (CV11) | A | 100 | 100 |
| 13. | Nigeria (CV9) | B.1 | 100 | 100 |
| 14. | Nigeria (CV8) | A | 100 | 100 |
| 15. | Nigeria (CV5) | A | 100 | 100 |
| 16. | Nigeria (CV4) | A | 100 | 99 |
| 17. | Nigeria (CV3) | B.2.1 | 100 | 98 |
A - Root of the pandemic lies within lineage A. Many sequences originating from China and many global exports; including to South East Asia, Japan, South Korea, Australia, the USA and Europe, represented in this lineage
B.1 - A large European lineage that corresponds to the Italian outbreak.
B.2.1 - Large lineage with representation from UK, Europe, Jordan, Australia, USA, India, Ghana (Bootstrap=11)
**Quick information about the lineages (obtained from https://github.com/hCoV-2019/lineages ) [Rambaut et al ., 2020]
Genome annotation and Mutation Analysis
We investigated the presence of the globally spread spike protein mutation D614G which has steadily increased temporally in Europe, East coast of the United States, South America, Africa and some parts of Asia [Korber et al., 2020], and found four [4] Nigerian patients infected with the Spike D614G mutant virus [Fig. 2]. Three of these patients presented with very severe disease. This mutation has been associated with higher transmission and pathogenicity, and also helps the virus to evade immune interventions as it dominates the wild type whenever it is introduced into a new location [Fig. 3]. We were able to successfully annotate 11 out of 20 genomes using prokka [Seemann, 2014] with the SARS-CoV-2 reference genome [NC_045512.2] as guide, and then extracted the spike glycoprotein [S gene] using a custom Bash script from individual genomes. Multiple sequence alignment of these S genes was done with MAFFT [Katoh et al ., 2002] and visualised on UGENE [Okonechnikov et al ., 2012.]
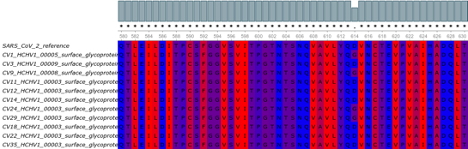
Figure 2: Multiple sequence alignment of the spike glycoprotein showing four [4] D614G spike mutations in the Nigerian SARS-CoV-2 genomes.
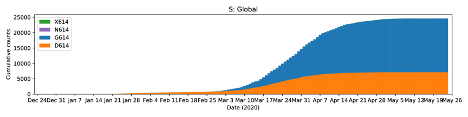
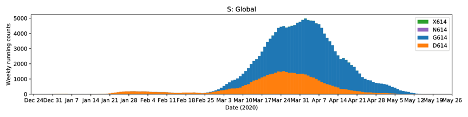
Figure 3: Global SARS-CoV-2 genomes [n=24,620] showing cumulative counts [top] and weekly running counts [bottom] of the D614G mutation [Korber et al., 2020] [cov.lanl.gov].
References
Katoh, K., Misawa, K., Kuma, K., & Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic acids research , 30 (14), 3059–3066. https://doi.org/10.1093/nar/gkf436.
Korber B., WM Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Foley B., Giorgi E.E., Bhattacharya T., Parker M.D., Partridge D.G., Evans C.M., Freeman T.M., de Silva T.I., on behalf of the Sheffield COVID-19 Genomics Group, LaBranche C.C., Montefiori D.C. (2020). Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2. bioRxiv 2020.04.29.069054; doi: https://doi.org/10.1101/2020.04.29.069054.
Matranga, C. B., Gladden-Young, A., Qu, J., Winnicki, S., Nosamiefan, D., Levin, J. Z., & Sabeti, P. C. (2016). Unbiased Deep Sequencing of RNA Viruses from Clinical Samples. Journal of visualized experiments : JoVE , (113), 54117. https://doi.org/10.3791/54117.
Okonechnikov K., Golosova O., Fursov M., the UGENE team (2012). Unipro UGENE: a unified bioinformatics toolkit, Bioinformatics , 28(8), 1166–1167, https://doi.org/10.1093/bioinformatics/bts091.
Price, M. N., Dehal, P. S., & Arkin, A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Molecular biology and evolution , 26 (7), 1641–1650. https://doi.org/10.1093/molbev/msp077
Rambaut A., Holmes E.C., Hill V., O’Toole A., McCrone J.T., Ruis C., du Plessis L., Pybus O.G. (2020). A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. bioRxiv 2020.04.17.046086; doi: https://doi.org/10.1101/2020.04.17.046086
Seemann T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics (Oxford, England) , 30 (14), 2068–2069. https://doi.org/10.1093/bioinformatics/btu153.
Data availability
All sequences are available at GitHub - acegid/CoV_Sequences: SARS-CoV-2 genome from Nigeria
GISAID, NCBI GenBank, and NCBI SRA accession numbers will be shared when available. We would like to thank all the authors who have kindly deposited and shared genome data on GISAID. A table with genome sequence acknowledgments can be found at GitHub - acegid/CoV_Sequences: SARS-CoV-2 genome from Nigeria.
Partners and Collaborators
African Centre of Excellence for Genomics of Infectious Diseases (ACEGID), Redeemer’s University, Ede, Osun State, @acegid .
Redeemer’s University, Ede, Osun State (RUN).
Nigeria Centre for Disease Control (NCDC), Abuja, Nigeria, @NCDCgov .
Africa CDC, Addis Ababa, Ethiopia, @AfricaCDC .
Broad Institute and Harvard University, Cambridge, MA, USA.
Beth Israel Deaconess Medical Center, Boston, MA, USA.
College of Medicine, University of Ibadan, Ibadan, Nigeria.
Disclaimer and contact information
Please note that these analyses are based on work in progress and should be considered preliminary. Our analyses of this data are ongoing and a publication communicating our findings on these and other published genomes is in preparation. These data cannot be used without permission. If you wish to use this data please contact:
Christian Happi, PhD
Professor of Molecular Biology and Genomics, Redeemer’s University, Ede, Osun State, Nigeria
Director, African Center of Excellence for Genomics of Infectious Diseases [ACEGID]
E-mail: [email protected]
Website: www.acegid.org
Twitter: @christian_happi
Chikwe Ihekweazu, M.P.H, F.F.P.H
Director General, Nigerian Centre for Disease Control [NCDC], Abuja, Nigeria
Email: [email protected]
Website: [https://ncdc.gov.ng/]
Twitter: @Chikwe_I
Paul Eniola Oluniyi, (PhD in view)
African Center of Excellence for Genomics of Infectious Diseases (ACEGID)
Redeemer’s University, Ede, Osun State, Nigeria
E-mail: [email protected]
Twitter: @pauloluniyi
Idowu Olawoye, (PhD in view)
African Center of Excellence for Genomics of Infectious Diseases (ACEGID)
Redeemer’s University, Ede, Osun State, Nigeria
Email: [email protected]
Twitter: @idowuolawoye