SARS-CoV-2 evolution, post-Omicron
Cornelius Roemer1, Ryan Hisner2, Nicholas Frohberg2, Hitoshi Sakaguchi3, Federico Gueli4, Thomas P. Peacock5
1Biozentrum, University of Basel, Basel, Switzerland; Swiss Institute of Bioinformatics
2Independent Researcher, USA
3Retired, Japan
4Independent Researcher, Italy
5Department of Infectious Disease, Imperial College London, UK
*corresponding author: [email protected]
Summary
Following the emergence and global spread of Omicron lineage BA.5, there has been an unprecedented diversification of Omicron sublineages. In this report we describe the different evolutionary trajectories and mechanisms driving the emergence of these sublineages and give examples of the most prevalent lineages in each, covering topics such as; second generation BA.2 lineages, simple and complex recombination, antigenic drift and convergent evolution.
Introduction
Since the emergence of the Omicron variant in late 2021, many regions have experienced repeated waves of infections driven by successive Omicron lineages, for example BA.1, BA.2, then BA.5. BA.1-5 are all hypothesised to have directly arisen from the same cryptic ‘Omicron reservoir’(1, 2). At present, a mix of Omicron sub-variants are emerging and growing, often coinciding with waves of infections. Here we aim to give a summary of the evolutionary mechanisms through which these new ‘post-Omicron’ lineages are emerging.
Saltation/Second generation variants
In the years since its emergence, SARS-CoV-2 has shown a apparently unique evolutionary feature relative to what is known for other common respiratory viruses – the ability to produce ‘variants’ defined by long phylogenetic branch lengths, an apparent lack of genetic intermediates and rooting within older, rather than contemporary lineages(1-3). Interestingly this may have parallels with the evolution of pandemic norovirus strains(4). This ‘saltation’ or ‘variant’ evolution is hypothesised to be the result of the re-emergence of viruses evolved during long-term chronic infections (1, 5-7). The extended duration of chronic infections may allow the virus to more quickly accumulate mutations due to the lack of a transmission bottleneck. Examples of probable saltation variants include the variants of concern (VOCs) Alpha, Beta, Gamma and the Omicron lineages (e.g. BA.1, BA.2 and BA.5)(1, 3, 8). Notably all these original variants evolved from pre-variant progenitors.
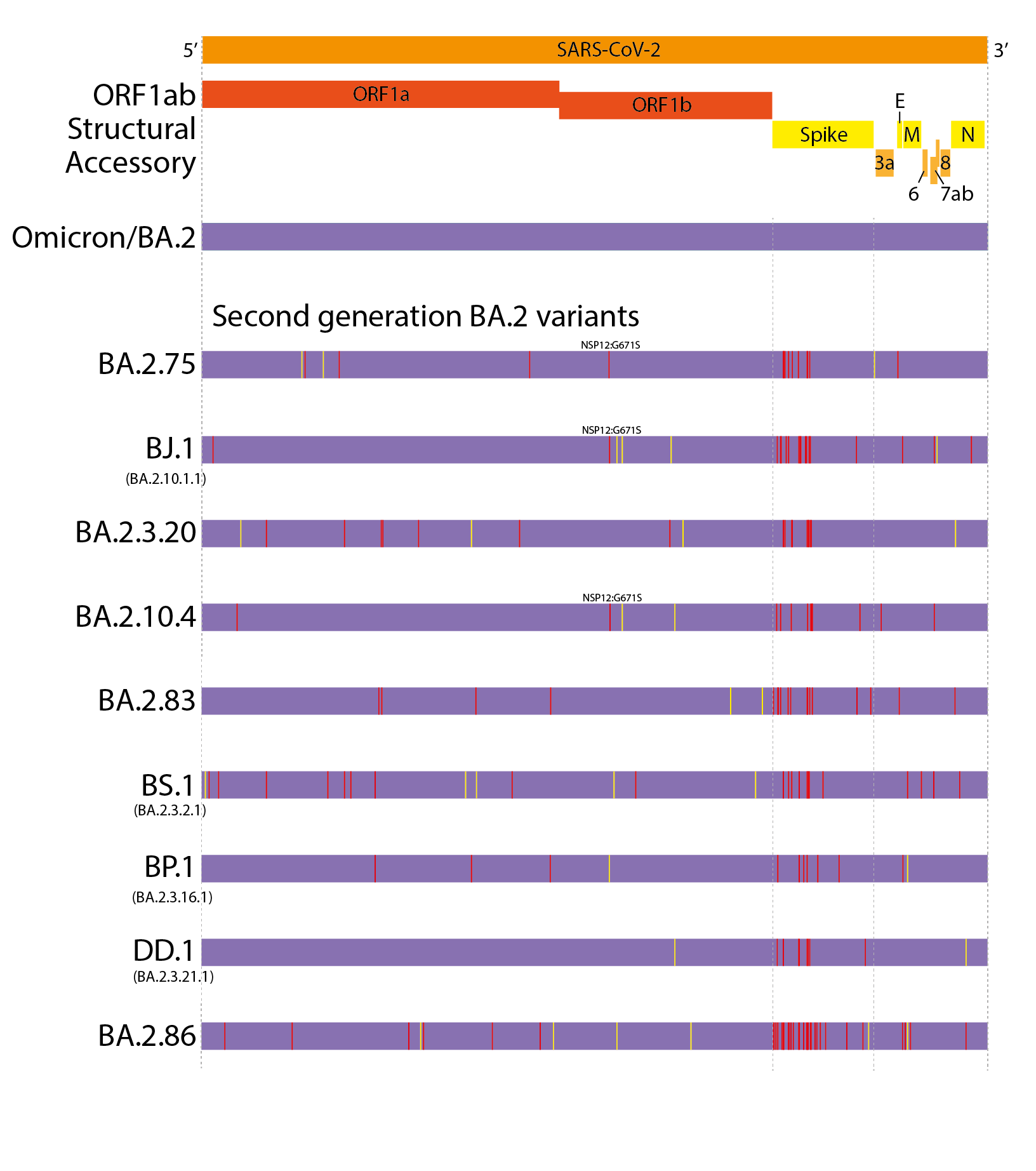
This evolutionary pattern has continued into 2022. However, unlike the first generation variants, these have largely evolved from a BA.2 background, resulting in ‘second generation variants’ - variants that have arisen from an existing variant background (Figure 1 and 2). Examples of this include lineages such as BA.2.75, BA.2.10.4, BJ.1, BS.1(9), BA.2.3.20, BA.2.83, BP.1, and DD.1. Like prior first generation variants, these lineages have numerous non-synonymous mutations, particularly concentrated in the N-terminal domain (NTD) and receptor binding domain (RBD) of the Spike (Figure 1)(10). Although worldwide sequencing efforts have been declining throughout 2022, it still appears there are no genetic intermediates sampled between these variants and their precursors (Figure 2), suggesting they too may have evolved from chronic BA.2 infections, seeded near the end of 2021 or start of 2022.
At present, BA.2.75 is by far the most widespread of the second generation BA.2 variant lineages, though this is partially due to it arising first among these lineages. BA.2.3.20 has also shown some appreciable growth in recent months.
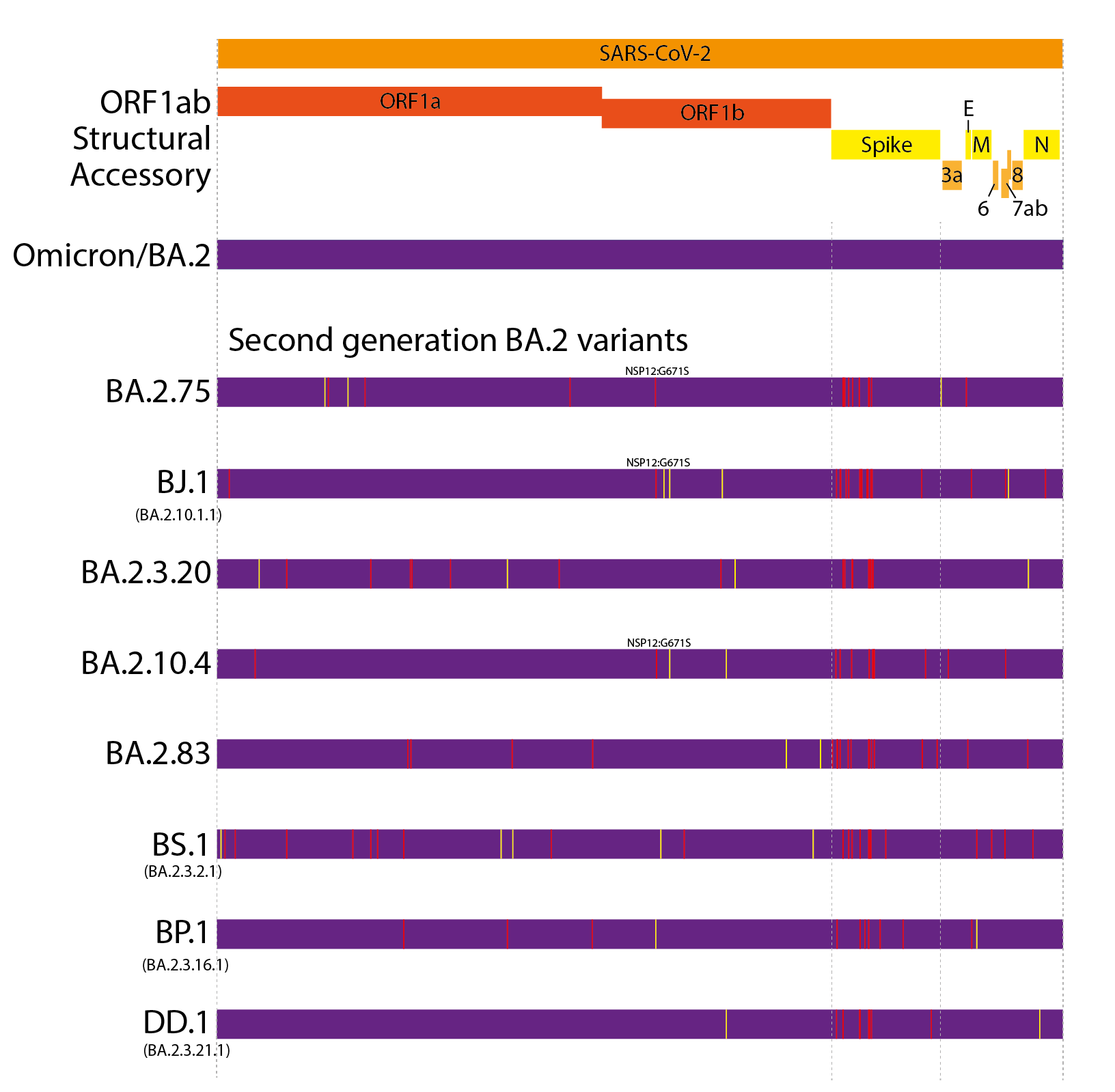
Figure 1. Defining mutations of the saltation/second generation BA.2 variant lineages. Synonymous or non-coding mutations shown in yellow, non-synonymous mutations shown in red. Non-spike mutations showing convergent evolution annotated onto genomes.

Figure 2. Phylogenetic relatedness and convergent evolution in contemporary lineages. Phylogenetic tree modified from nextclade curated tree with select fast growing or large convergent lineages(11). Table shows the most prominent convergent positions. Grey boxes indicate the site is the same as BA.2. Coloured boxes indicate substitutions at that site. Light labeled boxes indicate new mutations on branch, dark boxes indicate substitution shared with the parental lineage. Recombinant lineages shown as dotted lines.
‘Simple’ recombination
Recombination is common in coronaviruses. Ever since there has been enough genetic diversity to unambiguously identify chimeric genomes, it has been well established SARS-CoV-2 is highly prone to inter-lineage recombination(12). Generally emergence of recombinants have come when a prior wave is in steep decline and the next variant is already emerging(12). This has invariably led to these recombinant lineages being outcompeted alongside their parental lineages. However, it is well recognised that recombinants between divergent variants can contain advantageous properties unique to both parents(13)… At present (Late November 2022) there are 54 Pango-designated recombinant lineages, denoted by their X- prefix (14).
XBB is the most widespread inter-lineage recombinant to date (11). XBB is a recombinant between two saltation BA.2 lineages, BJ.1 and a BA.2.75 sublineage - most likely BM.1.1.1 (Figure 3). XBB inherited the 5’ part of its genome from BJ.1 and the 3’ end of its genome from BA.2.75, with a single breakpoint within the RBD of Spike. This Spike breakpoint allows it to possess potent antigenic RBD mutations from both BJ.1 and BA.2.75, leading it to have one of the most potent combinations of antigenic RBD mutations of any circulating variant, resulting in a relatively large antigenic distance from prior variants(15). Other notable contemporary recombinant lineages are XBD and XBF, both of which are recombinants between BA.5 and BA.2.75 sublineages (Figure 3).
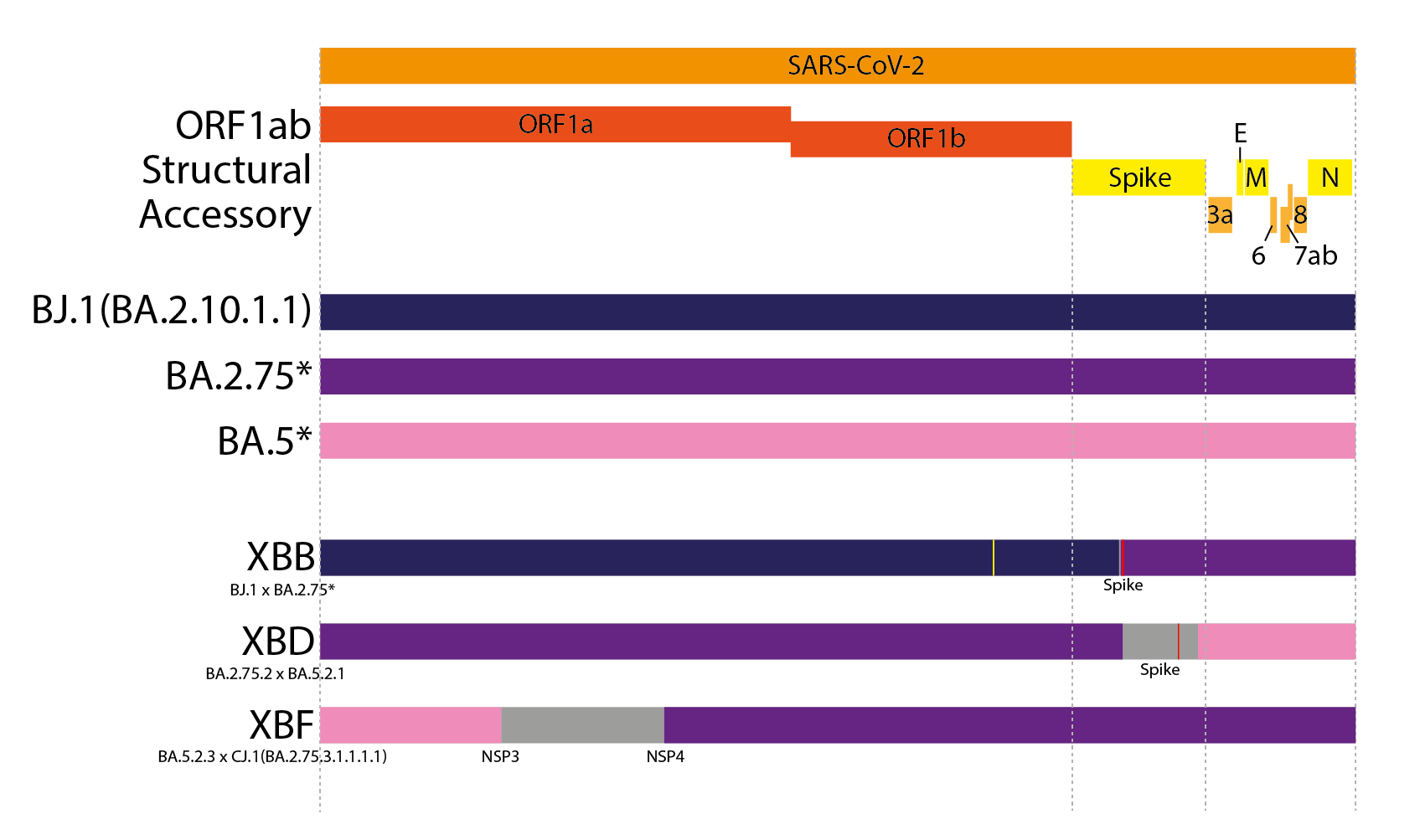
Figure 3. Genome schematics of the contemporary ‘simple’ recombinant lineages XBB, XBD and XBF. Different colours indicate different parts of the genome from each parent. Grey areas indicate ambiguous areas that most likely contain the breakpoint. Red lines indicate non-synonymous private mutations while yellow lines indicate synonymous private mutations.
‘Complex’ recombination
Beyond the ‘simple’ recombinants, a novel evolutionary trend has been seen in 2022, the emergence of ‘complex’ recombinants. Prior SARS-CoV-2 recombinants have been the result of recombinant events between extant, co-circulating lineages, and have tended to be the result of 1 or 2 detectable breakpoints. Complex recombinants instead are between lineages that did not widely co-circulate (in several cases Delta and BA.2), often contain far greater numbers of breakpoints (3-8), and also contain much higher numbers of ‘private mutations’ (mutations in the genome that do not appear to come from either parental lineage) compared to ‘simple’ recombinants (Figure 4). XAY and XBA, furthermore, share parts of their genomes and private mutations with one another, suggesting they likely arose from the same source(16). Other examples of complex recombinants are XAW (which has only 2 breakpoints) and, most recently, XBC.
Due to the sometimes high number of breakpoints and private mutations, and the non-contemporary parental lineages, we hypothesise that these ‘complex’ recombinants, like the saltation variants, have emerged from chronic infections. In the case of XAY, XBA, XBC, and XAW, these likely arose from chronic infections where the individual was initially infected by Delta and subsequently superinfected with BA.2.
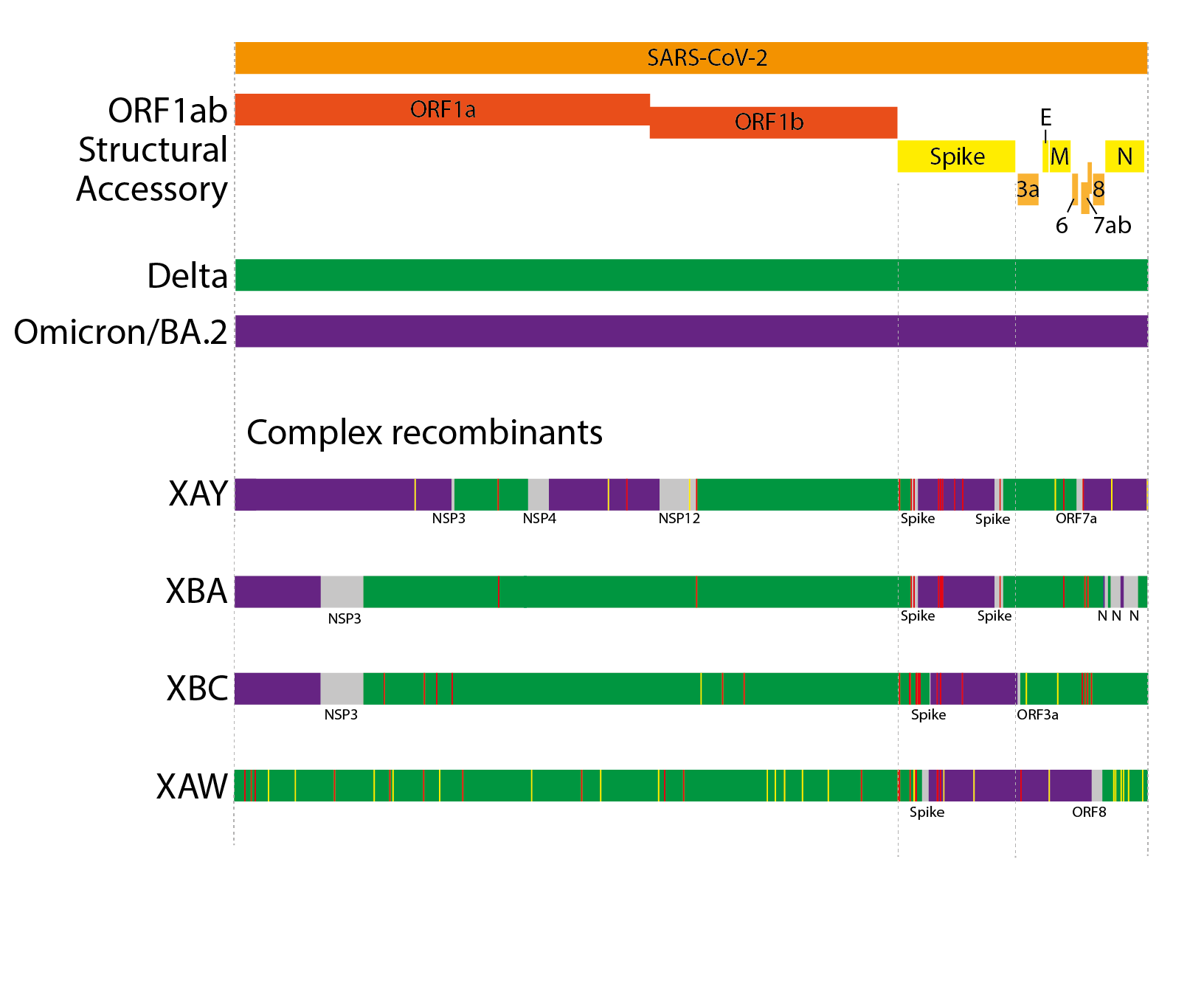
Figure 4. Schematics of the genome organisations of complex Delta x BA.2 recombinants. Coloured areas indicate sections most likely from one parent or another. Grey areas indicate ambiguous areas that likely contain the breakpoints. Red lines indicate non-synonymous private mutations while yellow lines indicate synonymous private mutations.
Of these complex recombinants XBC and XAY appear the most widespread. However both these lineages contain fewer antigenic RBD mutations than rapidly growing lineages such as BQ.1.1 or XBB (Figure 2), and therefore may be unlikely to compete in the long term.
Drift
By mid 2022, BA.5 had become the predominant variant globally, displacing BA.2 in most regions. Unlike previous dominant lineages, which showed relatively little accumulation of antigenic mutations once they predominated (with the closest examples being BA.2.12.1 or BA.1.1), BA.5 instead began to accumulate potent antigenic mutations in a step-wise manner - this is as opposed to the initial BA.2 sublineage saltation variants which appear to accumulate these mutations ‘all at once’. The most rapidly growing example of such lineages are sublineages of BQ.1, the largest of which is BQ.1.1, which contains 3 further antigenic mutations in its spike receptor binding domain (RBD) – the main target for neutralising antibodies in SARS-CoV-2 (Figure 2). Several other examples, with less extensive mutations have also shown recent growth, such as BA.4.6, BF.7 and BQ.1.1’s parental lineage, BQ.1(11).
Furthermore, some of the aforementioned second generation variants derived from BA.2 also show similar antigenic drift. In particular BA.2.75 has generated a huge number of sublineages that have accumulated antigenic RBD mutations by a more step-wise antigenic drift-like mechanism (Figure 2). Notable examples include BA.2.75.2, BR.2, BN.1.2.1, BM.1.1.1 and CH.1.1 all of which contains several additional antigenic RBD mutations compared to parental BA.2.75(11, 15). Overall this ‘drift’-like evolutionary pattern is much more in line with other respiratory viruses, for example step-wise antigenic drift in seasonal influenza viruses or HCoV-229E(17, 18).
Convergent evolution
One final feature common to all the previously discussed variants is the high degree of convergent evolution, particularly at antigenic RBD sites(15). Substitutions such as R346X, K444X, G446X, L452X, N460K, F486X (particularly F486P, a 2 nucleotide change), F490X and the R493Q reversion (to name a few) are present in many of these saltation, drift and recombinant lineages. Additionally several NTD changes, particularly deletions in the ~144 region are also appearing on multiple branches(15). One hypothesis for why these same sites are being selected for in so many different lineages is that these are key antigenic mutations - or possibly ACE binding enhancing sites compensating for antigenic mutations in the case of N460K and R493Q. It is thought that humoral immune responses primed with the ancestral strains (through vaccination or prior infection) and then boosted by Omicron infection specifically generates a dominant response that targets epitopes these sites reside within(15). This antigenic exposure leads to the enrichment of antibodies targeting sites that are conserved between the ancestral and Omicron lineages and these specific sites have therefore come under high levels of selective pressure leading to this observed convergent evolution (Figure 2). It is unclear if mutations such as these will continue to accumulate over time at further, less dominant sites, or whether these mutations will slow down due to fitness costs associated with further mutations.
The future of SARS-CoV-2 evolution
At present BQ.1.1, XBB and CH.1.1 appear to be the fastest growing variants globally, and are expected to drive some level of waves of infections in the coming months, either together or individually. Although these lineages contain substitutions at the same antigenic RBD residues, often the exact substitutions differ (Figure 2) - the 3 variants also have very different combinations of substitutions and deletions in their NTDs. Therefore, it is feasible immune responses elicited by these lineages may poorly cross-react with one another. Co-circulation of SARS-CoV-2 variants has generally been transient up to the present but it is possible several lineages could have similar enough growth rates, and enough antigenic distance from one another that they co-circulate, at least until a fitter lineage or variant emerges.
At present it is one year since the emergence of Omicron(1). Although all the lineages described here have genetic and antigenic distance from earlier Omicron lineages, they are still likely to continue to display somewhat similar viral, epidemiological and clinical properties to their parent lineages. However, it is entirely possible all the lineages described here would be outcompeted in the event of a second ‘Omicron-like event’ – the emergence of a brand new variant with orthogonal antigenicity from any other circulating lineages, for example from an ancestral or pre-Omicron variant genetic background. Due to its singular nature, it is extremely unclear how likely or how commonly we should expect such events going forwards, however it seems prudent to have strategies in place in the event this were to occur.
Emergence of a saltation variant from a Delta genetic background is of particular concern, although it’s entirely possible the intrinsically higher pathogenicity (19) of Delta might well have changed upon re-emergence. It should be noted that a number of Delta sequences continue to be sampled, most with large numbers of private mutations suggesting there is an existing, potentially substantial, reservoir of Delta (as well as prior variants). Such sequences most likely represent ongoing chronic infections, as exemplified by the emergence of the complex recombinants.
To conclude, it is essential for genomic surveillance and analysis of SARS-CoV-2 to continue even while other pandemic measures are phased out. As described throughout this report, the virus is continuing to evolve at pace by both predictable as well as hard to predict mechanisms, often at the same time. Equitable global surveillance, in particular will be essential for rapid responses to new variants and is currently very lacking and globally uneven (20).
Acknowledgements
We gratefully acknowledge all data contributors, i.e., the Authors and their Originating laboratories responsible for obtaining the specimens, and their Submitting laboratories for generating the genetic sequence and metadata and sharing via the GISAID Initiative (21), on which this research is based. The analysis in this study is based on the 1,578,488 sequenced collected after 2022-07-01 available on GISAID as of 2022-11-25, via EPI_SET_221201bv, accession available at doi: 10.55876/gis8.221201bv. Supplementary table available here: gisaid_supplemental_table_epi_set_221201bv.pdf - Google Drive
The authors would also like to thank the Pango designation committee and wider contributors for identifying and naming lineage, and the teams behind Nextstrain, covSPECTRUM, and UShER for developing tools that are used to identify and surveil lineages. The authors would also like to thank Daniel Sheward of the Karolinska Institutet, Sweden, and Shay Fleishon for their valuable feedback and discussions. T.P.P is funded by the MRC funded G2P-UK National Virology Consortium (MR/W005611/1).
References
-
Viana R, Moyo S, Amoako DG, Tegally H, Scheepers C, Althaus CL, et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature. 2022.
-
Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature. 2021;592(7854):438-43.
-
Hill V, Du Plessis L, Peacock TP, Aggarwal D, Colquhoun R, Carabelli AM, et al. The origins and molecular evolution of SARS-CoV-2 lineage B.1.1.7 in the UK. Virus Evolution. 2022;8(2).
-
Ruis C, Lindesmith LC, Mallory ML, Brewer-Jensen PD, Bryant JM, Costantini V, et al. Preadaptation of pandemic GII.4 noroviruses in unsampled virus reservoirs years before emergence. Virus Evolution. 2020;6(2).
-
Kemp SA, Collier DA, Datir RP, Ferreira IATM, Gayed S, Jahun A, et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature. 2021;592(7853):277-82.
-
Wilkinson SAJ, Richter A, Casey A, Osman H, Mirza JD, Stockton J, et al. Recurrent SARS-CoV-2 mutations in immunodeficient patients. Virus Evolution. 2022;8(2).
-
Harari S, Tahor M, Rutsinsky N, Meijer S, Miller D, Henig O, et al. Drivers of adaptive evolution during chronic SARS-CoV-2 infections. Nature Medicine. 2022.
-
Tegally H, Moir M, Everatt J, Giovanetti M, Scheepers C, Wilkinson E, et al. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nature Medicine. 2022.
-
Japanese National Institute of infectious Diseases (NIID). 感染・伝播性の増加や抗原性の変化が懸念される 新型コロナウイルス(SARS-CoV-2)の変異株について (第20報). www.niid.go.jp; 2022.
-
Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM, et al. SARS-CoV-2 variants, spike mutations and immune escape. Nature Reviews Microbiology. 2021;19(7):409-24.
-
Sheward DJ, Kim C, Fischbach J, Sato K, Muschiol S, Ehling RA, et al. Omicron sublineage BA.2.75.2 exhibits extensive escape from neutralising antibodies. The Lancet Infectious Diseases. 2022;22(11):1538-40.
-
Jackson B, Boni MF, Bull MJ, Colleran A, Colquhoun RM, Darby AC, et al. Generation and transmission of interlineage recombinants in the SARS-CoV-2 pandemic. Cell. 2021;184(20):5179-88.e8.
-
Simon-Loriere E, Montagutelli X, Lemoine F, Donati F, Touret F, Bourret J, et al. Rapid characterization of a Delta-Omicron SARS-CoV-2 recombinant detected in Europe. Research Square. 2022.
-
Pybus O. Pango Lineage Nomenclature: provisional rules for naming recombinant lineages. Virologicalorg. 2021.
-
Cao Y, Jian F, Wang J, Yu Y, Song W, Yisimayi A, et al. Imprinted SARS-CoV-2 humoral immunity induces converging Omicron RBD evolution. bioRxiv. 2022:2022.09.15.507787.
-
(NGS NfGSiSA, SA). SARS-CoV 2 Sequencing Update 15 July 2022. NGS-SA report; 2022.
-
Smith DJ, Lapedes AS, de Jong JC, Bestebroer TM, Rimmelzwaan GF, Osterhaus ADME, et al. Mapping the Antigenic and Genetic Evolution of Influenza Virus. Science. 2004;305(5682):371-6.
-
Eguia RT, Crawford KHD, Stevens-Ayers T, Kelnhofer-Millevolte L, Greninger AL, Englund JA, et al. A human coronavirus evolves antigenically to escape antibody immunity. PLoS Pathog. 2021;17(4):e1009453.
-
Twohig KA, Nyberg T, Zaidi A, Thelwall S, Sinnathamby MA, Aliabadi S, et al. Hospital admission and emergency care attendance risk for SARS-CoV-2 delta (B.1.617.2) compared with alpha (B.1.1.7) variants of concern: a cohort study. The Lancet Infectious Diseases. 2022;22(1):35-42.
-
Brito AF, Semenova E, Dudas G, Hassler GW, Kalinich CC, Kraemer MUG, et al. Global disparities in SARS-CoV-2 genomic surveillance. Nature Communications. 2022;13(1):7003.
-
Shu Y, McCauley J. GISAID: Global initiative on sharing all influenza data – from vision to reality. Eurosurveillance. 2017;22(13):30494.