RASCL: Rapid assessment of SARS-COV-2 clades enabled through molecular sequence analysis and its application to B.1.617.1 and B.1.617.2
Alexander G Lucaci1, Jordan D Zehr1, Stephen D Shank1, Dave Bouvier2, Han Mei2, Anton Nekrutenko2, Sergei L Kosakovsky Pond1
1Institute for Genomics and Evolutionary Medicine, Temple University, Philadelphia, Pennsylvania, USA
2Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA, USA
The ongoing circulation and diversification of SARS-CoV-2 has spawned a large number of genotypically distinct viral lineages, with various geographic distributions, different infectivities, and varying levels of immune resistance (Hamed et al., 2021, Young et al., 2021). An odds-on expectation is that new viral lineages will continue to emerge, either by further evolution of parental lineages, or “jump” events (e.g. intra-host evolution for the B.1.1.7 lineage) for the foreseeable future (Rambaut et al., 2020). Certain lineages or clades have been designated variants of interest (VOI) or variants of concern (VOC), to highlight important changes in their phenotypes, or the acquisition of mutations that are known to abrogate antibody binding or reduce vaccine efficacy (Luchsinger et al, 2021, Abdool et al., 2021). A viral lineage that has acquired a set of key distinguishing mutations may continue evolving under existing or shifting selective pressures, and it is of clear interest to identify where and how these selective forces may be targeting viral genomes.
In order to monitor the continued evolution of the SARS-CoV-2 genome, we developed a rapid assessment tool designed to investigate the nature and extent of selective forces acting on viral genes in clade sequences. Our application relies on performing comparative phylogenetic analyses to identify clade-specific molecular evolution by evaluating selective pressures for ‘query’ clade sequences against automatically selected background sequences. This design offers the ability to rapidly interrogate SARS-CoV-2 clades for evidence of pervasive, episodic, or directional selection as well as reporting on physicochemical properties of substitutions where appropriate. In addition, we report on several characteristic indicators including the extent of diversifying, positive, purifying, and coevolutionary pressures. The results of our application are presented through the use of interactive notebooks (Perkel 2021).
Results
We investigated the nature and extent of selective forces acting on the viral genes in both B.1.617.1 and B.1.617.2 clade sequences. Sequence collection, sampling, and deposition into public data repositories is an ongoing process. Analyses are performed on data pulled from GISAID (Elbe et al., 2017) on May 11, 2021. Using complete linkage distance clustering (https://github.com/veg/tn93) we selected a median of 67 B.1.617.1 and 59 B.1.617.2 sequences from available sequences to represent genomic diversity. A global reference dataset from publicly available sequences via ViPR (Pickett et al., 2012) is included as background.
For both clades, we find strong statistical support for multiple modes of selection: diversifying positive (FEL/MEME), negative (FEL), and directional (FADE). There is also evidence of coevolution among sites within individual gene segments (BGM), and at a number of sites clade evolution occurs with selective forces that are different from those acting on background sequences (Contrast-FEL). These results are summarized in Figures 1 and 2 and can be fully explored in the following interactive notebook: https://bit.ly/3oXB65l.
A key observation is that selection appears to be operating on many sites that are not a part of the clade-defining signature mutation set, implying that there may be ongoing diversification and adaptation in these clades.

Figure 1. Schematic for genome-level results for the B.1.617.1 clade. The color key is defined as follows: Green = MEME (episodic diversifying selection) on All branches (Track 1, or the outermost track) and MEME results on Internal branches (Track 2). Purple = Clade defining sites from Pangolin. Orange = S1 Functional sites shown to influence antibody binding. Yellow = Contrast-FEL results on Internal branches. Blue = FADE results, a test for directional selection. Grey box = FEL results for negatively selected sites. Grey lines = BGM results for coevolving sites.

Figure 2. Schematic for the genome-level results for the B.1.617.2 clade. The color key is defined as follows: Green = MEME results on All branches (Track 1, or the outermost track) and MEME results on Internal branches (Track 2). Purple = Clade Defining sites from Pangolin. Orange = S1 Functional sites shown to modify antibody binding. Yellow = Contrast-FEL results on Internal branches. Blue = FADE results, a test for directional selection. Grey box = FEL results for negatively selected sites. Grey lines = BGM results for coevolving sites.
We next focus our attention on positively selected sites located in the Spike gene. We identify 21 sites subject to episodic positive selection in the B.1.617.1 clade (Table 1), with only three of those appearing on the canonical clade signature list (484, 681, 1071). Evolutionary patterns for most of these sites suggest that multiple selective events occurred, i.e. repeated substitutions to the same residue across multiple tree branches. Several sites have also been shown, individually or in combination with others to reduce mAb or convalescent/vaccine plasma efficacy (https://covdb.stanford.edu). Five of the sites also increased in frequency from the period preceding March 15th, to the period following March 15th (>2x fold change). The trends in most common haplotypes at those five positions are shown in Figure 3. Additional three sites (157, 22, 859) did not have any mutations prior to March 15th, but acquired mutations following that date (Figure 4).
| Genomic Coordinate | Mutation | MEME # branches under selection | MEME q-value | Fold change : pre-/post-March 15 | Notes |
|---|---|---|---|---|---|
| 21568 | V3G | 2 | 0.032 | 2.3 | Directional: G |
| 21616 | T19R | 2 | 0.0533 | 19.9 | |
| 21844 | T95I | 8 | 1.11e-08 | 1 | mAb / plasma reduction in combination |
| 21985 | G142D | 11 | 5.18e-09 | 0.8 | mAb / plasma reduction in combination |
| 22021 | E154K | 7 | 0.00048 | 1.1 | mAb / plasma reduction in combination |
| 22030 | F157S | 1 | 0.0824 | None before March 15 | |
| 22213 | Q218H | 2 | 0.0497 | 1.1 | |
| 22225 | A222V | 1 | 0.0868 | None before March 15 | |
| 22705 | V382L | 2 | 0.0844 | 0.5 | |
| 23011 | E484Q | 3 | 0.0319 | 0.9 | mAb / plasma reduction |
| 23275 | T572SI | 1 | 0.0857 | None before March 15 | |
| 23404 | V615I | 1 | 0.0846 | ||
| 23602 | P681HR | 2 | 0.0376 | 1 | |
| 23677 | A706V | 1 | 0.0874 | 0.3 | |
| 24136 | T859N | 1 | 0.0861 | None before March 15 | |
| 24388 | S943PT | 2 | 0.05 | 3.6 | |
| 24409 | D950NH | 3 | 0.00256 | 5.3 | |
| 24772 | Q1071H | 5 | 0.00261 | 0.9 | |
| 24862 | H1101DY | 2 | 0.0128 | 0.8 | |
| 25120 | N1187H | 2 | 0.0314 | 1.1 | |
| 25351 | V1264L | 1 | 0.0818 | 3.2 | Directional: L |
| Table 1. Sites in B.1.617.1 which show evidence for episodic selection. Sites which are bolded indicate clade-defining mutations from Pangolin. |
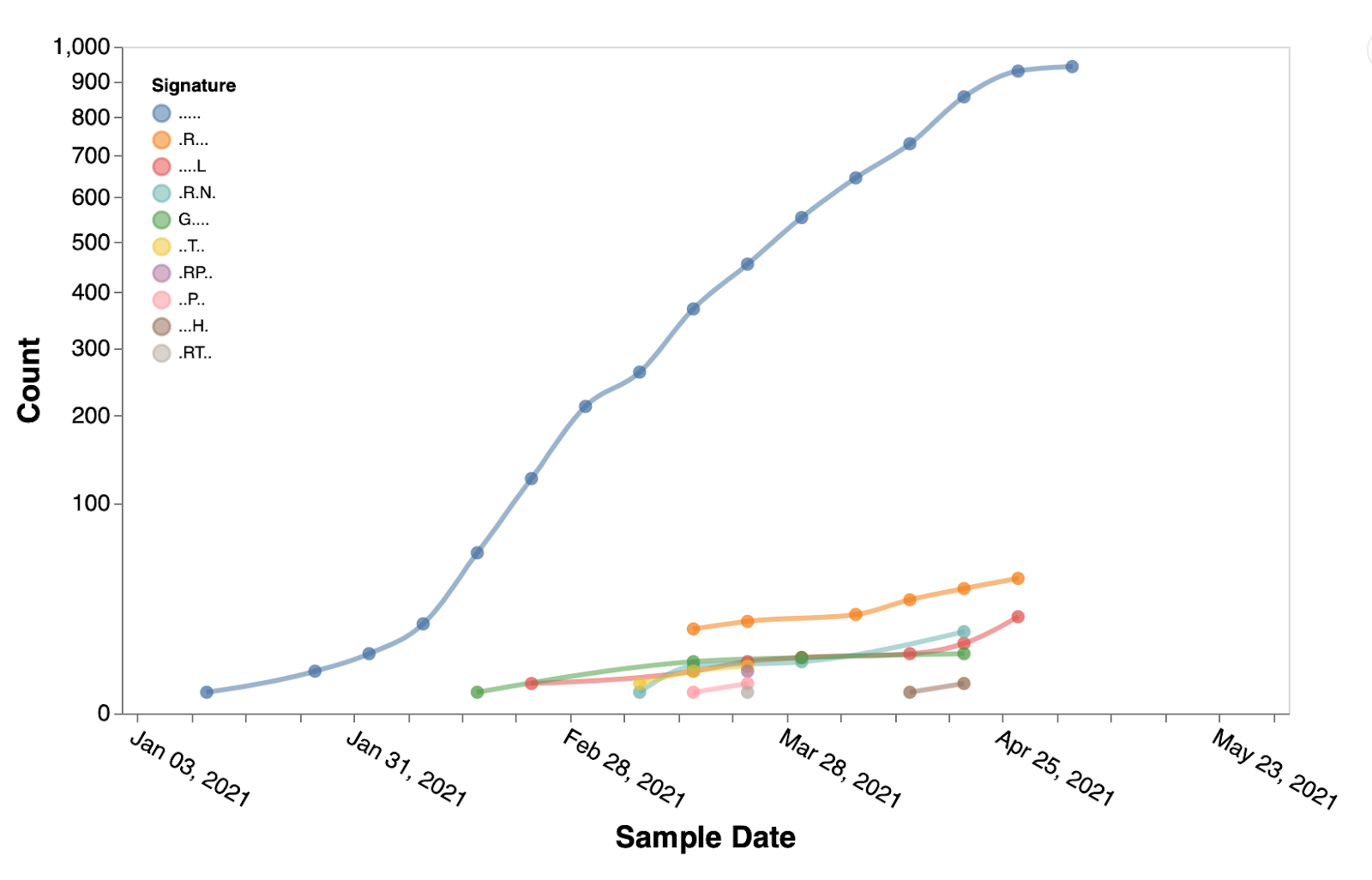
Figure 3. Temporal trends of the substitution combinations at sites 3, 19, 943, 950, 1264 in B.1.617.1 sequences.
. denotes the reference residue. Figures like this can be generated using Trends in mutational patterns across SARS-CoV-2 genes enabled by data from / Sergei Pond | Observable
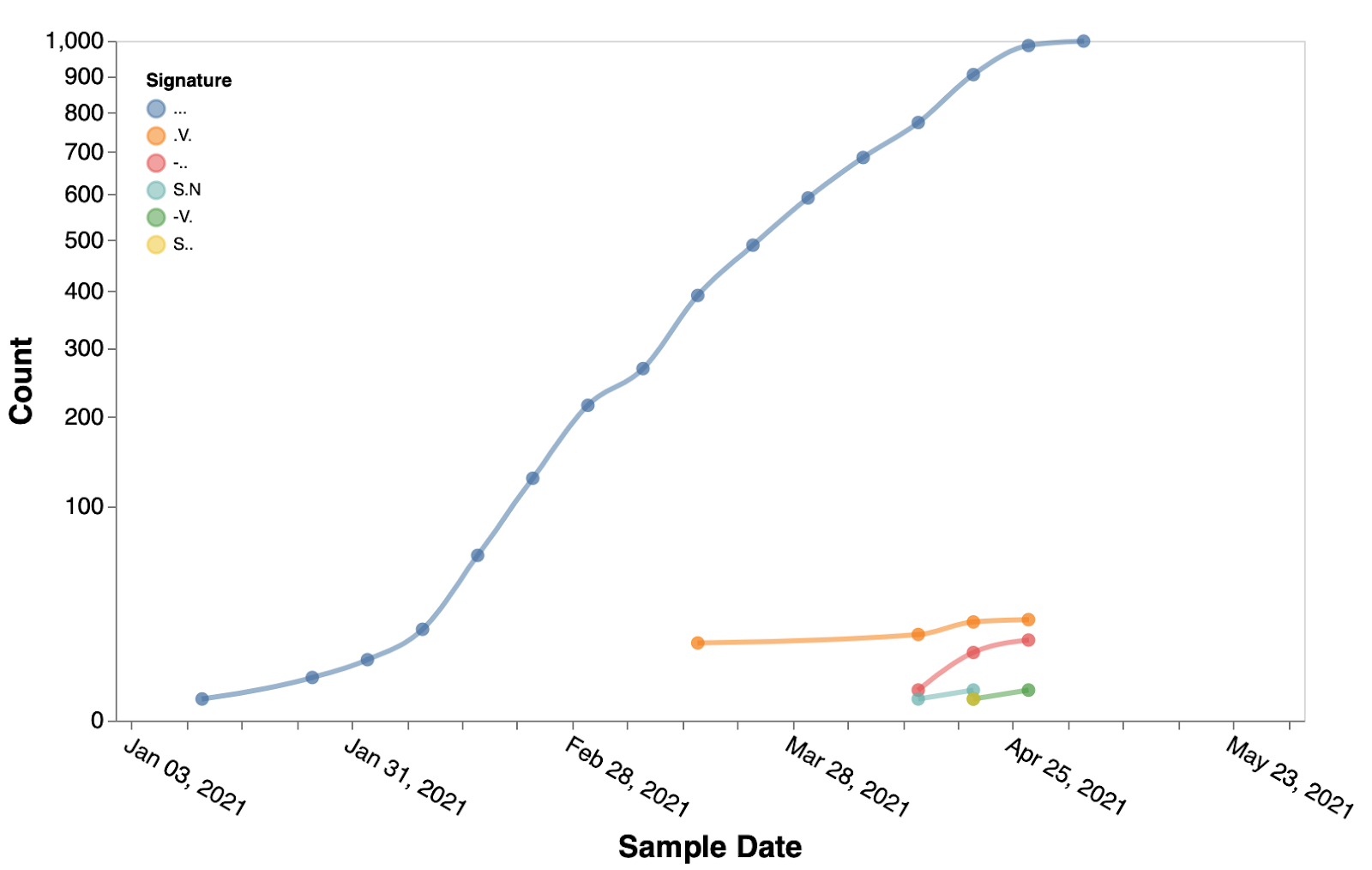
Figure 4. Temporal trends of the substitution combinations at sites 157, 222, 859 in B.1.617.1 sequences.
. denotes the reference residue.
| Genomic Coordinate | Mutation | MEME # branches under selection | MEME q-value | Fold change : pre-/post-March 15 | Notes |
|---|---|---|---|---|---|
| 21574 | L5F | 2 | 0.00829 | Not seen before March 15 | Directional F |
| 21844 | T95I | 2 | 0.00487 | 8.7 | |
| 21985 | G142D | 5 | 0.000231 | 1.0 | Directional D. mAb epitope binding |
| 22225 | A222V | 4 | 0.000123 | 0.8 | Directional V |
| 23281 | D574YH | 1 | 0.0139 | Not seen before March 15 | |
| 24409 | D950N | 3 | 0.000928 | 1.0 | |
| 25351 | V1264L | 1 | 0.0322 | Not seen before March 15 |
Table 2. Sites in B.1.617.2 which show evidence for episodic selection. Sites which are bolded indicate clade-defining mutations from Pangolin.
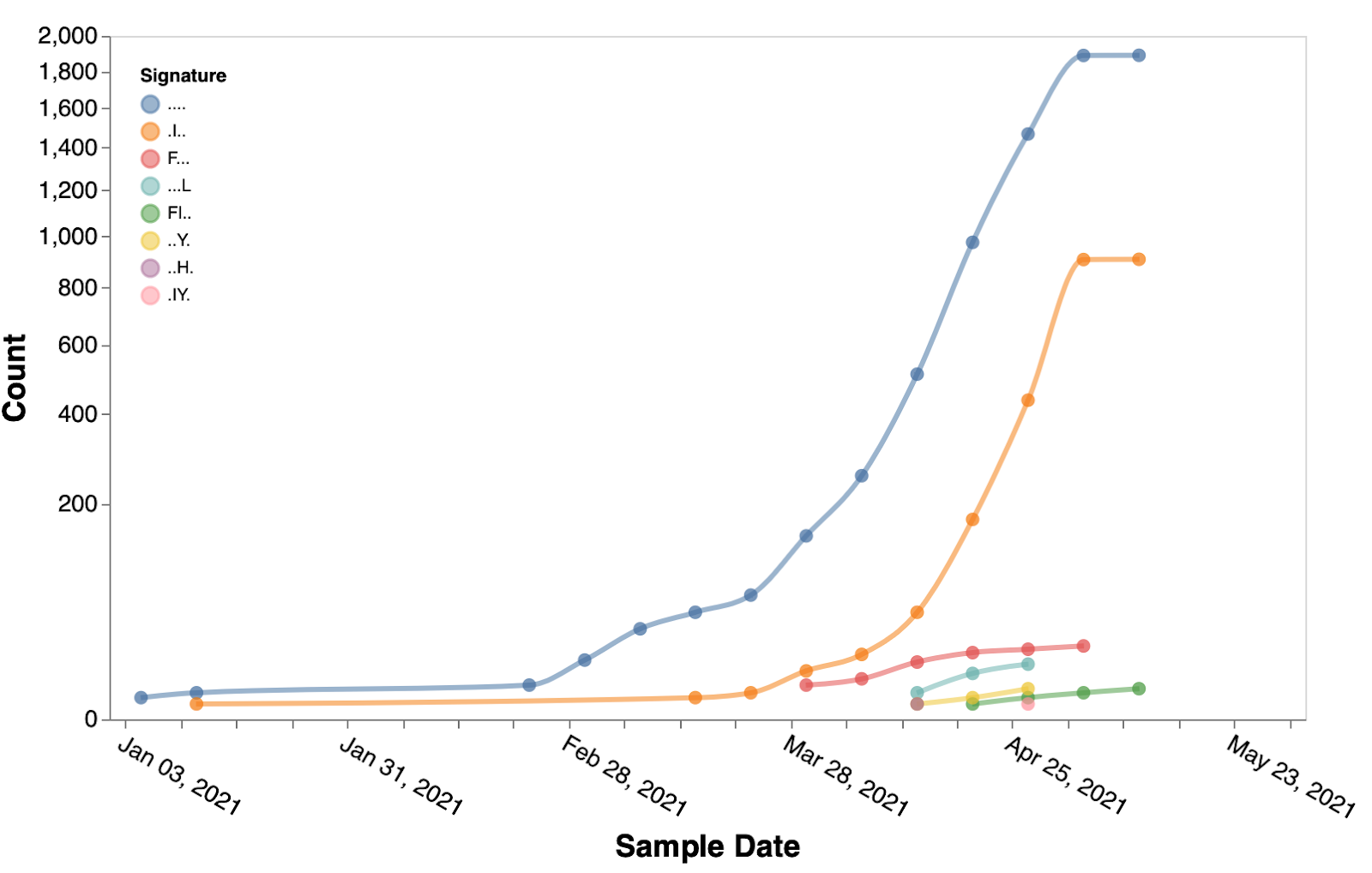
Figure 5. Temporal trends of the substitution combinations at sites 5, 95, 574, 1264 in B.1.617.2 sequences.
. denotes the reference residue.
Methods
The RASCL application is designed to accept two inputs: a “query” whole genome sequence dataset, and a “background” whole genome sequence dataset. Users are requested to input a whole genome dataset (fasta, unaligned), which will be defined as the “query” sequence dataset, and can be typically retrieved from data repositories such as GISAID. Typically, a background sequence dataset is assembled from publicly available sequences via the ViPR database, and is provided by default unless otherwise specified. However, our application also tolerates the usage of another clade of interest to be used as “background”, which allows one to directly interrogate between-clade evolutionary dynamics.
Whole genome datasets are broken down into their respective coding sequences from the NCBI SARS-CoV-2 reference annotation. The gene set includes structural (Spike, Matrix, Nucleocapsid, Envelope) and non-structural (leader, nsp2, nsp3, nsp4, 3C, nsp6, nsp7, nsp8, nsp9, nsp10, helicase, exonuclease, endornase, ORF3a, ORF6, ORF7a, ORF8, RdRp, methyltransferase) genes. Individual genes from query and background datasets are processed from here onwards. Our procedure includes several quality control steps including sanitizing fasta input headers and striking ambiguous codons from an alignment. In order for our analyses to complete rapidly, alignments are subsampled using genetic distances provided by the TN93 (Tamura and Nei 1993) algorithm. A combined (query + background) alignment is created and with sequences kept from the background dataset divergent enough to be useful for subsequent selection analysis. Inference of a maximum likelihood phylogenetic tree (RAxM-NG, Kozlov et al., 2019) is performed on the combined dataset. Trees are labelled for “query” and “background” branches. Analyses SLAC (Pond and Frost, 2005), BGM (Poon et al., 2008), FEL (Pond and Frost, 2005), MEME (Murrell et al., 2012), aBSREL (Smith et al., 2015), BUSTEDS (Wisotsky et al., 2020), RELAX (Wertheim et al., 2015), CFEL (Pond et al., 2021) are performed with state of the art molecular evolution models from the HyPhy (Pond et al., 2020) suite of bioinformatics tools. We report on sites of interest which together may represent regions of ongoing evolution within a specific clade. Results are combined into easily readable and web-accessible JSON files used for web processing. Visualization of results is done with full-feature interactive notebooks through ObservableHQ.
Software availability RASCL is available from https://github.com/veg/SARS-CoV-2_Clades.
Acknowledgements
We are thankful for the GISAID contributors from across the world. Additionally, we thank members of the Datamonkey (Weaver et al., 2020) / HyPhy and Galaxy teams for their assistance in the development of this application.
References
- Elbe, S., and Buckland-Merrett, G. (2017) Data, disease and diplomacy: GISAID’s innovative contribution to global health. Global Challenges, 1:33-46. DOI:10.1002/gch2.1018 PMCID: 31565258
- Pickett BE, Sadat EL, Zhang Y, Noronha JM, Squires RB, Hunt V, Liu M, Kumar S, Zaremba S, Gu Z, Zhou L, Larson CN, Dietrich J, Klem EB, Scheuermann RH. ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012 Jan;40(Database issue):D593-8. doi: 10.1093/nar/gkr859. Epub 2011 Oct 17. PMID: 22006842; PMCID: PMC3245011.
- Sergei L Kosakovsky Pond, Art FY Poon, Ryan Velazquez, Steven Weaver, N Lance Hepler, Ben Murrell, Stephen D Shank, Brittany Rife Magalis, Dave Bouvier, Anton Nekrutenko, Sadie Wisotsky, Stephanie J Spielman, Simon DW Frost, Spencer V Muse (2020) HyPhy 2.5—A Customizable Platform for Evolutionary Hypothesis Testing Using Phylogenies. Molecular Biology and Evolution 37.1 (2020): 295-299.
- Steven Weaver, Stephen D. Shank, Stephanie J. Spielman, Michael Li, Spencer V. Muse, Sergei L. Kosakovsky Pond Datamonkey 2.0: a modern web application for characterizing selective and other evolutionary processes Mol. Biol. Evol. 35(3):773–777
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993 May;10(3):512-26. doi: 10.1093/oxfordjournals.molbev.a040023. PMID: 8336541.
- Alexey M Kozlov, Diego Darriba, Tomáš Flouri, Benoit Morel, Alexandros Stamatakis, RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference, Bioinformatics, Volume 35, Issue 21, 1 November 2019, Pages 4453–4455, RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference | Bioinformatics | Oxford Academic
- Sergei L. Kosakovsky Pond and Simon D. W. Frost (2005) Not So Different After All: A Comparison of Methods for Detecting Amino Acid Sites Under Selection Molecular Biology and Evolution 22(5): 1208-1222
- Art F. Y. Poon, Fraser I. Lewis, Simon D. W. Frost, Sergei L. Kosakovsky Pond, Spidermonkey: rapid detection of co-evolving sites using Bayesian graphical models, Bioinformatics, Volume 24, Issue 17, 1 September 2008, Pages 1949–1950, https://doi.org/10.1093/bioinformatics/btn313
- Ben Murrell, Joel O. Wertheim, Sasha Moola, Thomas Weighill, Konrad Scheffler and Sergei L. Kosakovsky Pond (2012) Detecting Individual Sites Subject to Episodic Diversifying Selection PLoS Genetics 8(7): e1002764
- M. D. Smith, J. O. Wertheim, S. Weaver, B. Murrell, K. Scheffler and S. L. Kosakovsky Pond Less Is More: An Adaptive Branch-Site Random Effects Model for Efficient Detection of Episodic Diversifying Selection Molecular Biology and Evolution 32: 1342-1353
- Wisotsky SR, Kosakovsky Pond SL, Shank SD, Muse SV. Synonymous Site-to-Site Substitution Rate Variation Dramatically Inflates False Positive Rates of Selection Analyses: Ignore at Your Own Peril. Mol Biol Evol. 2020 Aug 1;37(8):2430-2439. doi: 10.1093/molbev/msaa037. PMID: 32068869; PMCID: PMC7403620.
- Wertheim JO, Murrell B, Smith MD, Kosakovsky Pond SL, Scheffler K. RELAX: detecting relaxed selection in a phylogenetic framework. Mol Biol Evol. 2015 Mar;32(3):820-32. doi: 10.1093/molbev/msu400. Epub 2014 Dec 23. PMID: 25540451; PMCID: PMC4327161.
- Kosakovsky Pond SL, Wisotsky SR, Escalante A, Magalis BR, Weaver S. Contrast-FEL-A Test for Differences in Selective Pressures at Individual Sites among Clades and Sets of Branches. Mol Biol Evol. 2021 Mar 9;38(3):1184-1198. doi: 10.1093/molbev/msaa263. PMID: 33064823; PMCID: PMC7947784.
- Hamed, S.M., Elkhatib, W.F., Khairalla, A.S. et al. Global dynamics of SARS-CoV-2 clades and their relation to COVID-19 epidemiology. Sci Rep 11, 8435 (2021). https://doi.org/10.1038/s41598-021-87713-x
- Association of SARS-CoV-2 clades with clinical, inflammatory and virologic outcomes: An observational study. Young, Barnaby E et al. EBioMedicine, Volume 66, 103319
- Rambaut, A., Loman, N., Pybus, O., Barclay, W., Barrett, J., Carabelli, A., … & Volz, E. (2020). Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. Genom. Epidemiol.
- Luchsinger LL, Hillyer CD. Vaccine efficacy probable against COVID-19 variants. Science. 2021 Mar 12;371(6534):1116. doi: 10.1126/science.abg9461. PMID: 33707257.
- Abdool Karim SS, de Oliveira T. New SARS-CoV-2 Variants - Clinical, Public Health, and Vaccine Implications. N Engl J Med. 2021 May 13;384(19):1866-1868. doi: 10.1056/NEJMc2100362. Epub 2021 Mar 24. PMID: 33761203; PMCID: PMC8008749.
- Perkel JM. Reactive, reproducible, collaborative: computational notebooks evolve. Nature. 2021 May;593(7857):156-157. doi: 10.1038/d41586-021-01174-w. PMID: 33941927.