Thomas P. Peacock1*, David L. V. Bauer2, Wendy S. Barclay1
1Department of Infectious Disease, St Marys Medical School, Imperial College London, UK.
2The RNA Virus Replication Laboratory, The Francis Crick Institute, London, UK.
*corresponding author – [email protected]
Summary
SARS-CoV-2 continues to evolve and adapt to humans. In this report, we describe RNA insertions, particularly those in the SARS-CoV-2 Spike protein, and show how they mostly cluster in the Spike N-terminal domain and at the S1/S2 cleavage site. While many insertion sequences appear to be viral in origin, we find that a subset of insertions show homology to RNA sequences from host transcripts, implying incorporation of short host RNA sequences during viral genome replication.
Introduction
Over the course of the COVID-19 pandemic, SARS-CoV-2 has accumulated mutations that likely further adapt it to humans. These mutations have occurred through three major mechanisms – RNA substitutions, deletions and, less commonly, insertions. Insertions have been less well characterised but are of particular interest due to i) being less ‘random’ than deletions/substitutions as they require a source of the novel RNA sequence and ii) being of particular interest to the origin of SARS-CoV-2, which has a unique 12 nucleotide insert in its Spike protein, resulting in the creation of a furin cleavage site.
Several variant lineages contain insertions in their spike proteins, most notably the variant of interest, Mu (or B.1.621) which contains a 3 nucleotide insertion in its aa~140 region of Spike, as well as the A.2.5 and B.1.214.2 variants which both have insertions in the aa~210 insertion hotspot region (Gerdol, Dishnica et al. 2021). In addition to circulating variants, insertions have also been identified in chronic infections of individuals (Hoffman, Costales et al. 2021), and upon serial passage of SARS-CoV-2 in cell culture (Andreano, Piccini et al. 2021, Shiliaev, Lukash et al. 2021).
In this report we investigate the potential origins of both widespread and rare insertions in the SARS-CoV-2 genome, and relate this back to what is known about genome insertions in other RNA viruses.
Results
Spike insertions are mostly found in the N-terminal domain and around the S1/S2 cleavage site.
The Spike protein of SARS-CoV-2 shows the greatest amount of variation amongst circulating strains. Mutations in Spike are associated with changes in SARS-CoV-2 transmissibility, pathogenicity and other viral properties. To investigate the distribution of insertions into the SARS-CoV-2 Spike protein, we searched for spike insertions using GISAID and manually removed sequences that appeared to be most obviously artefactual (See Method section). We found that Spike insertions mostly fall within the Spike N-terminal domain, or in the S1/S2 furin cleavage site region. Both of these regions are also known hotspots for deletions (McCarthy, Rennick et al. 2021), further confirming their plasticity.
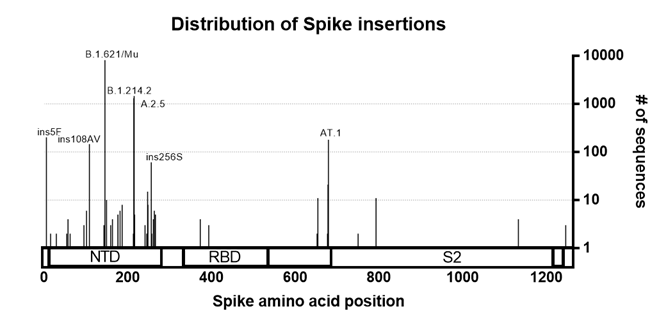
Figure 1. Distribution of insertions in the Spike protein. Sequences with insertion in the Spike were pulled from GISAID (as of 8th October 2021). Sequences were curated to remove low quality/likely artefacts and plotted against the Spike protein sequence. Lineages of interest and insertions with high numbers of sequences associated are annotated.
Many nucleotide insertions in SARS-CoV-2 appear to be viral in origin
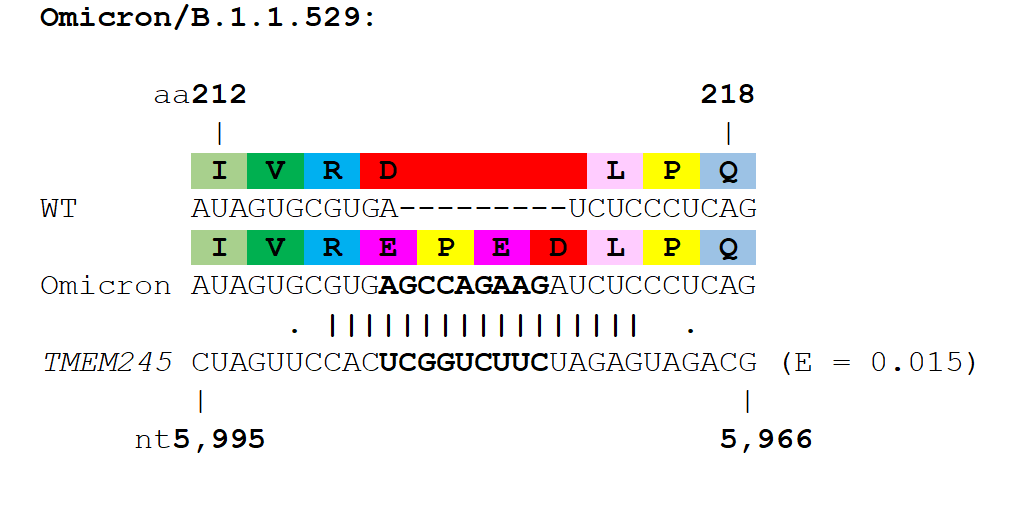
Next, we investigated the potential origins of the insertion sequences, and expanded the analysis to several non-Spike insertions. Many insertions are clearly duplications of adjacent codons or codon pairs. For example, a recurrent Spike ins678QT insertion, encoded as CAG ACT, has been seen arising independently across multiple SARS-CoV-2 lineages. Another subset of insertions show high homology to distal parts of the SARS-CoV-2 genome, often in the opposite sense (see Table 1 and Figure 2), indicating as others have suggested, that they likely result from copy-choice recombination from the template genome during replication (Garushyants, Rogozin et al. 2021). One notable example is the Russian AT.1 lineage, which contains a 12-nucleotide insertion at the S1/S2 site: the AT.1 insertion shows high homology to the 3’UTR of the SARS-CoV-2 genome which is present in viral genomes and all sub-genomic RNAs, and therefore abundant within viral replication organelles.
| Insertion | Pango lineage | Monophyletic? | Location | Gisaid IDs | Putative region of origin | E value |
|---|---|---|---|---|---|---|
| Spike ins654GAEGALNTP | B.1.617.2 | 2 clusters | India | 7 x sequences | N 28,661-28,708 | 6.00E-09 |
| Spike ins159INTTC | B.1.2 cluster | Yes | Oregon, USA | EPI_ISL_1791466; EPI_ISL_1791465; EPI_ISL_1791473 | Spike 21,650-21,682 | 4.00E-06 |
| Spike ins214KAFKQ | B.1.1.7 cluster | Yes | Czech Republic | EPI_ISL_2228123; EPI_ISL_2228125 | NSP2 1745-1780 | 4.00E-05 |
| NSP3 ins153ENPHL | B.1.2 cluster | Yes | USA | 60x sequences | NSP12 15256-15294 | 4.00E-05 |
| Spike ins214KKLIRGD | B.1.638 cluster | Yes | South Africa | EPI_ISL_3451093; EPI_ISL_3451094 | Spike 22,745-22789 | 8.00E-05 |
| Spike ins214W | B.1 cluster | Yes | New York | EPI_ISL_4096626; EPI_ISL_4096639 | N 29,218-29,247 | 2.00E-04 |
| Spike ins261DGSDK/A262S | B.1.617.2 cluster | No | India | EPI_ISL_2461980; EPI_ISL_2461981 | NSP3 7,666-7,707 | 8.00E-04 |
| Spike ins214ANRN | B.1.1.28 cluster | Yes | South America | 7x sequences | M 26,631-26,660 | 0.002 |
| NSP4 ins265ICFA/N266Y | B.1.2 cluster | Mostly | USA | 9x sequences | NSP7 11,998-12,036 | 0.003 |
| Spike N679K/ins679GIAL | AT.1 lineage | Yes | Worldwide ex Russia | 174x sequences | 3’ UTR 29,820-29846 | 0.006 |
| Spike ins108AV | B.1.617.2 clusters | multiple clusters | Various | 108x sequences | NSP16 21,415-21,447 | 0.006 |
| ORF6 M58I/ins58FRSL | B.1.1.7 cluster | multiple clusters | Europe | 43x sequences | 5’ UTR 29-64 | 0.01 |
| Spike ins214QAS | B.1 cluster | Yes | North America | x19 sequences | N 1,156-1,182 | 0.017 |
| Spike ins214KRI | B cluster | Mostly | Denmark | EPI_ISL_972296; EPI_ISL_1880746; EPI_ISL_970811; EPI_ISL_929396 | NSP5 10,849-10,878 | 0.046 |
| Spike 247FKT | B.1.258.14 cluster | Yes | Italy | EPI_ISL_1229272; EPI_ISL_1359440; EPI_ISL_1359445; EPI_ISL_1558361 | NSP13 17,986-18,018 | 0.046 |
Table 1. Insertions with a high confidence of being viral (from distal sites implying template switching) in origin. Insertions shown in ascending order of BLASTN E values.
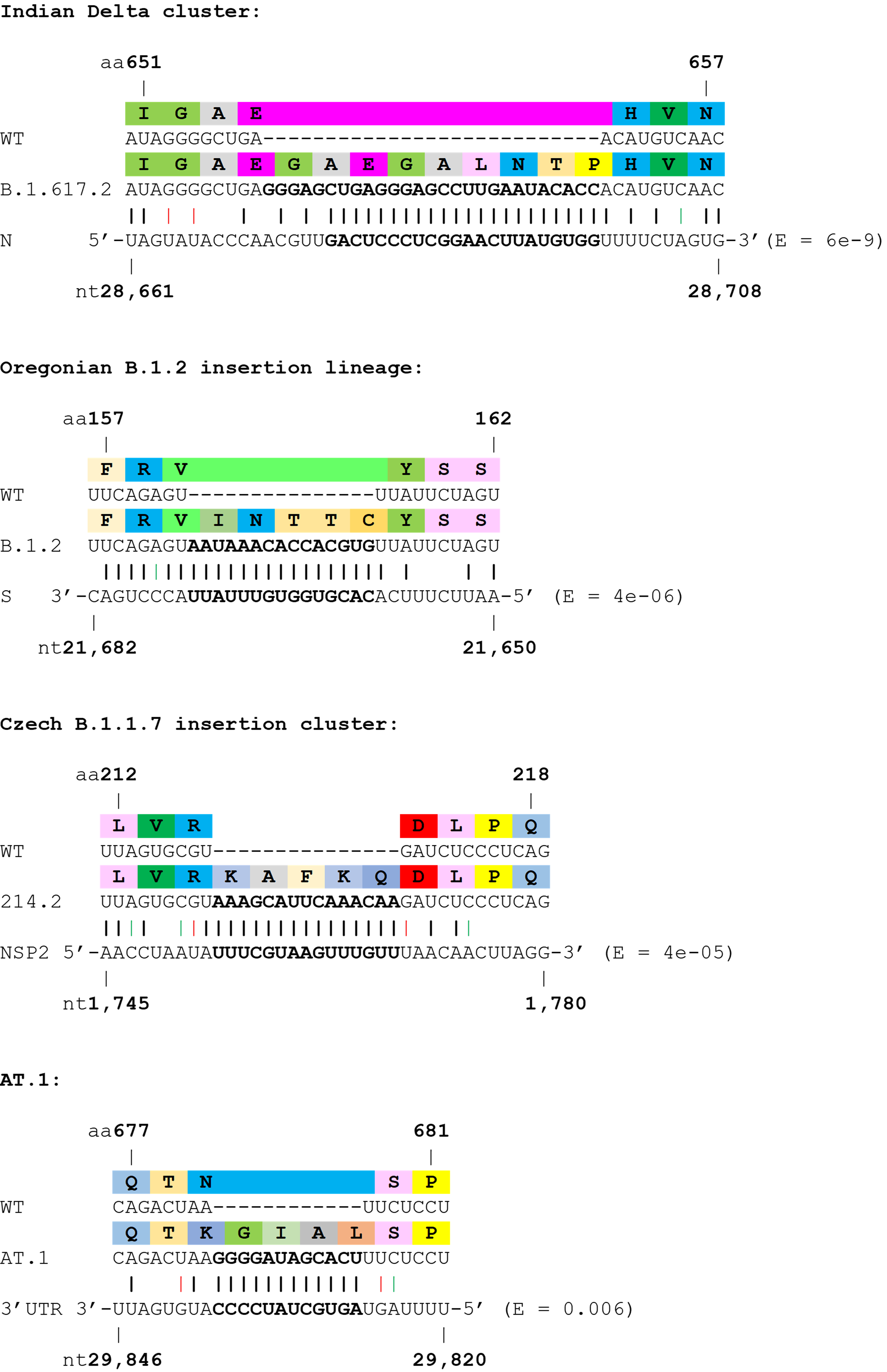
Figure 2. Examples of SARS-CoV-2 clusters or lineages with strong support for viral RNA template switching. Wobble base pairing shown in red (sense) or green (antisense). E values from BLASTN shown.
Several nucleotide insertions in SARS-CoV-2 appear to originate from host mRNAs.
Although an appreciable number of insertion sequences showed high similarity to proximal or distal regions of the SARS-CoV-2 genome, many did not. Therefore, we speculated that the other major source of RNAs that could be used as templates for insertions would be host mRNAs themselves. Therefore, we used BLASTN to look for homology between these RNA inserts and the host transcriptome. Several interesting hits were found with high homology to host mRNAs (see Table 2, Figure 3). For example, a cluster of B.1.1.519 viruses from the USA with a 12 nucleotide insertion in the N protein showed very high homology to human ZBTB20 transcript. Another highly relevant, but more tentative, example is the insertion seen in the variant of interest Mu/B.1.621, which shows a degree of homology to the mRNA of human TRIM28.
| Insertion | Pango lineage | Monophyletic? | Location | Gisaid IDs/references | Putative origin | E value |
|---|---|---|---|---|---|---|
| N ins390EMPV | B.1.1.519/B.1.1.7 clusters | No | West Virginia | EPI_ISL_2615927; EPI_ISL_2184872; EPI_ISL_2184874 | ZBTB20 9,337-9,372 | 5.00E-04 |
| ORF6 ins32TIIL | B.1.1.7 cluster | Yes | Scotland | EPI_ISL_1103220; EPI_ISL_1103106; EPI_ISL_1103136; EPI_ISL_1103154; EPI_ISL_1247705 | GOLIM4 4,194-4,226 | 0.011 |
| Spike ins214GLTSKRN | N/A | N/A | Laboratory mutant | Shiliaev et al | RASSF6 (Chlorocebus sabaeus) | 0.02 |
| Spike ins214KFH | B.1.1.7 cluster | Yes | Scotland | EPI_ISL_1190578; EPI_ISL_1063373; EPI_ISL_1123191 | HYDIN 9,463-9,492 | 0.029 |
| Spike ins216ADL | B.1.2 cluster | Yes | USA | EPI_ISL_1016594; EPI_ISL_1532440; EPI_ISL_1037192; EPI_ISL_1234589; EPI_ISL_2797236 | CTRC 139-168 | 0.029 |
| Spike ins214HSG | AY.4 cluster | Yes | UK | x7 sequences | PTPRB 4,412-4,444 | 0.029 |
| Spike ins214EGAE | AY.4 cluster | Yes | Germany | EPI_ISL_4223414; EPI_ISL_4223419 | ADIPOR1 18-50 | 0.029 |
| Spike Y144T/Y145S/ins145N | B.1.621 lineage | Yes | Worldwide ex Colombia | >5000 sequences | TRIM28 1,805-1,834 | 0.086 |
Table 2. Insertions with highest confidence of originating from the host. Insertions shown in ascending order of BLASTN E values.

Figure 3. Examples of SARS-CoV-2 clusters or lineages with support for host origin of RNA insertions. Wobble base pairing shown by full stops. Mismatches in the insertions shown in red. E values from BLASTN shown.
Final thoughts
Naturally occurring insertions of foreign RNA into viral genomes are rare but can have high consequence. Coronaviruses have previously incorporated genes from their hosts or other viruses. For example, the phosphodiesterases, NS2a of the Embecovirus subgenus (which includes OC43, HKU1 and MHV), and NS4b of MERS-CoV, are thought to be two independent acquisitions of vertebrate AKAP7. Furthermore, Embecovirus Haemagglutinin-Esterase glycoproteins share a high degree of structural homology to the same proteins from Orthomyxoviridae. However, in both these examples whole proteins or domains are used for a similar function to the host/other virus. Flaviviridae and Orthomyxoviridae have been shown to acquire host RNA sequences that confer phenotypes unrelated to their original function, more like the insertions proposed in this study. Firstly, the Pestivirus bovine viral diarrhea virus (BVDV) has been shown to repeatedly insert host RNA into its NS2 coding region which can alter polyprotein cleavage and lead to a cytopathic phenotype in cell culture. Interestingly, some cytopathic BVDV achieved the same phenotype using viral-derived RNA sequences. Finally, and perhaps most relevant to the origin of SARS-CoV-2, is the example of the 2012 Mexican highly pathogenic avian influenza H7N3 outbreak (Maurer-Stroh, Lee et al. 2013). Avian influenza viruses of the H7 and H5 subtypes can exist as either low or high pathogenicity depending on the presence of absence of a polybasic furin cleavage site in their haemagglutinin proteins. It has been proposed that the 2013 Mexican H7N3 virus gained its furin cleavage site from heterologous recombination with host 28S ribosomal RNA, thought to be possible due to genomic RNA replication in the host nucleus/nucleolus where nascent rRNA is synthesised.
Insertions in the SARS-CoV-2 genome are also of particular interest as they may have the potential for much greater phenotypic change than mutation or deletion alone - a prime example being the original insertion generating the furin cleavage site, which likely contributed to the pandemic potential of the virus. However, insertion of additional loops in the Spike NTD, or further insertions at the S1/S2 site may have the ability to change the antigenicity or cleavability of these regions, respectively and alter the phenotype of the viruses that emerge. These types of mutations therefore can act as ‘wildcard’ mutations that are hard to predict, and special care should be taken so they can accurately be identified and characterised.
Method
Spike insertions were identified using the search term “Spike_ins” in the GISAID database. Insertions were then curated confirming insertions were truly present and weren’t obviously artefacts using the following criteria: i) insertions were found in more than one sequence (i.e. were not unique); ii) insertions did not contain any unresolved nucleotides (i.e. N); iii) insertions were in frame and did not contain stop codons; iv) virus genomes did not contain multiple insertions; v) when sequences were aligned they still contained the insertions (i.e. were not artefacts from the metadata); vi) insertions had some degree of phylogenetic clustering (as shown using Usher) and, if they did not show much clustering, did not all come a from a single uploading laboratory and; vii) sequences that appeared to show cross-lineage contamination – for example the large number of Delta isolates with identical insertions and surrounding mutations to B.1.621/Mu which is more likely due to the corresponding sequencing tile, which is known to drop out in Delta with the ARTIC V3 primers, picking up low levels of Mu contamination, rather than true recombination/convergent evolution.
To assess whether insertions in SARS-CoV-2 showed homology to the viral genome or host transcriptome insertions and the flanking regions were assessed using BLASTN against a reference SARS-CoV-2 genome (NC_045512.2) and the refseq_select Homo sapiens database (or Chlorocebus sabaeus in the case of the insertion from Shiliaev et al (Shiliaev, Lukash et al. 2021)). E values from BLASTN were then reported with a cutoff of >0.05 (with select E values above 0.05 included for lineages of particular interest). To confirm Pango lineages and test whether insertion-containing clusters were monophyletic, sequences were analysed using Ultrafast Sample placement on Existing tRee (UShER; UCSC UShER: Upload)(Turakhia, Thornlow et al. 2021).
Acknowledgements
The authors would like to thank Dr Ada Yan and Dr Daniel Goldhill for their help with the analysis and drafting of this report and Professor Julian Hiscox, and other members of the UK-G2P for their invaluable insights into the mechanisms of viral replication and recombination.
References
Andreano, E., G. Piccini, D. Licastro, L. Casalino, N. V. Johnson, I. Paciello, S. Dal Monego, E. Pantano, N. Manganaro, A. Manenti, R. Manna, E. Casa, I. Hyseni, L. Benincasa, E. Montomoli, R. E. Amaro, J. S. McLellan and R. Rappuoli (2021). “SARS-CoV-2 escape from a highly neutralizing COVID-19 convalescent plasma.” Proceedings of the National Academy of Sciences 118(36): e2103154118.
Garushyants, S. K., I. B. Rogozin and E. V. Koonin (2021). “Insertions in SARS-CoV-2 genome caused by template switch and duplications give rise to new variants of potential concern.” bioRxiv: 2021.2004.2023.441209.
Gerdol, M., K. Dishnica and A. Giorgetti (2021). “Emergence of a recurrent insertion in the N-terminal domain of the SARS-CoV-2 spike glycoprotein.” bioRxiv: 2021.2004.2017.440288.
Hoffman, S. A., C. Costales, M. K. Sahoo, S. Palanisamy, F. Yamamoto, C. Huang, M. Verghese, D. A. Solis, M. Sibai, A. Subramanian, L. S. Tompkins, P. Grant, R. W. Shafer and B. A. Pinsky (2021). “SARS-CoV-2 Neutralization Resistance Mutations in Patient with HIV/AIDS, California, USA.” Emerg Infect Dis 27(10): 2720-2723.
Maurer-Stroh, S., R. T. C. Lee, V. Gunalan and F. Eisenhaber (2013). “The highly pathogenic H7N3 avian influenza strain from July 2012 in Mexico acquired an extended cleavage site through recombination with host 28S rRNA.” Virology Journal 10(1): 139.
McCarthy, K. R., L. J. Rennick, S. Nambulli, L. R. Robinson-McCarthy, W. G. Bain, G. Haidar and W. P. Duprex (2021). “Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape.” Science: eabf6950.
Shiliaev, N., T. Lukash, O. Palchevska, D. K. Crossman, T. J. Green, M. R. Crowley, E. I. Frolova and I. Frolov (2021). “Natural isolate and recombinant SARS-CoV-2 rapidly evolve in vitro to higher infectivity through more efficient binding to heparan sulfate and reduced S1/S2 cleavage.” bioRxiv: 2021.2006.2028.450274.
Turakhia, Y., B. Thornlow, A. S. Hinrichs, N. De Maio, L. Gozashti, R. Lanfear, D. Haussler and R. Corbett-Detig (2021). “Ultrafast Sample placement on Existing tRees (UShER) enables real-time phylogenetics for the SARS-CoV-2 pandemic.” Nature Genetics 53(6): 809-816.