Preliminary Results from two novel ARTIC-style amplicon based sequencing approaches for RSV A and RSV B
Daniel M Maloney1,2*; Goncalo Fernandes1*; Rebecca Dewar1; Aine O’Toole2; Chenge Mphanga3; Thomas C Williams3; Martin McHugh1; Andrew Rambaut2; Kate E Templeton1
* These authors contributed equally
1 - Viral Sequencing Service, Royal Infirmary of Edinburgh, UK
2 - Institute of Ecology and Evolution, University of Edinburgh, UK
3 - Child Life and Health, University of Edinburgh, UK
Contact: [email protected]
Introduction:
Here we introduce two novel amplicon-based primer schemes for the sequencing of respiratory syncytial virus (RSV) A and RSV B genomes. With the atypical seasonality of RSV since the onset of the SARS-CoV-2 pandemic, as well as the approaching start of immunisation against RSV, there has never been as much of a demand for high quality, easy to produce genomes. The SARS-CoV-2 sequencing effort has highlighted the utility and flexibility of short amplicon based sequencing approaches, such as those designed by the ARTIC network, to enable genomic surveillance at an unprecedented scale. As such, the two primer schemes presented here have been specifically designed to slot into existing SARS-CoV-2 sequencing infrastructure to enable the easy switching of methodologies for the now extensive network of viral genomic sequencing laboratories to high throughput RSV genomic surveillance. These preliminary data presented here demonstrate the ability of these amplicon-based primer schemes to produce genomes with an average completeness of >95% from samples at least up to Ct 30, for both RSV A and RSV B.
Methods:
Sample selection: RSV A or B positive samples from the NHS Lothian area (South East Scotland; with appropriate tissue bank approval; Lothian NRS BioResource RTB approval (REC ref – 20/ES/0061)) were identified via Luminex NxTag Respiratory Pathogen Panel testing. Ct values were then established via rt-PCR using the methodology outlined in Templeton et al., 2004. Samples with Ct <30 in this assay were selected for sequencing. In total 45 RSV A and 45 RSV B samples were chosen, ~50% of which were samples from the 2019/2020 RSV season with the remaining half from 2021/2022.
Primer scheme design: Initial work focused on selecting geographically diverse high quality RSV genomes from the GISAID database in order to design a tiled amplicon primer scheme. In total 6 RSV A and 6 RSV B genomes were selected, with collection dates spanning 2019 to 2021, and locations spanning the UK, Australia and the USA. Draft primer schemes were generated using Primal Scheme (GitHub - aresti/primalscheme: a tool for designing primer panels for multiplex PCR.; Quick et al., 2017). The most recent UK based sample was used as the primary reference for primal scheme design for each scheme. An amplicon size of 400bp was targeted, to facilitate a drop-in replacement to existing SARS-CoV-2 lab protocols, resulting in two distinct primer schemes containing 50 amplicons each, one for RSV A and another for RSV B.
Nanopore Sequencing: Briefly, libraries of 48 samples (45 clinical samples plus 3 negative controls) were prepared using the Artic Lo-Cost V3 protocol for SARS-CoV-2 sequencing with minor in house modifications (full SOPs can be acquired upon request). Final libraries were loaded on R9.4.1 flow cells following the manufacturers specifications and run for 16 hours.
Consensus sequence generation: We assessed negative controls and calculated overall coverage following the sequencing runs using RAMPART (GitHub - artic-network/rampart: Read Assignment, Mapping, and Phylogenetic Analysis in Real Time). Negatives passed if fewer than 20 reads mapped to the RSV genome. Consensus sequences were generated using an in-house version of the “field bioinformatics” pipeline (v1.2.1) for tiled amplicon consensus sequence generation (GitHub - artic-network/fieldbioinformatics: The ARTIC fieldbioinformatics pipeline) with a 20x depth threshold and using Nanopolish for variant calling. Phylogenetic analysis was run on all consensus genomes with less than 10% ambiguous content. A background dataset of all complete GISAID RSV A and RSV B sequences with less than 5% ambiguity was used, with sequences aligned via MAFFT v7 (MAFFT - a multiple sequence alignment program; Katoh et al., 2002) and a Maximum-Likelihood tree with the Jukes-Cantor model of substitution (Jukes and Cantor, 1969) was built using IQTree v1.6.12 (http://www.iqtree.org/; Minh et al., 2020). Resulting trees were rendered using baltic.
Results :
Initial sequencing results indicate a near complete success rate for all RSV A and B samples sequenced to date. Of a total of 45 clinical samples for RSV A, 39 (86.7%) showed genome completeness above 90%, with a median genome completeness across all RSV A samples of 96.6%. Results were similarly positive for RSV B samples and 39 (86.7%) out of 45 clinical samples showed genome completeness above 90%, with a median genome completeness of 97.2% (Figure 1). Samples were preselected for Ct values below 30, and there was no discernible drop in genome completeness as Ct value increased up to 30 (Figure 2). Read counts for each sample did vary, however the minimum number of reads required for a sample to reach genome completeness of 90% remained low for both primer schemes at 28,662 mapped reads for RSV A and 35,447 for RSV B (Figure 2).
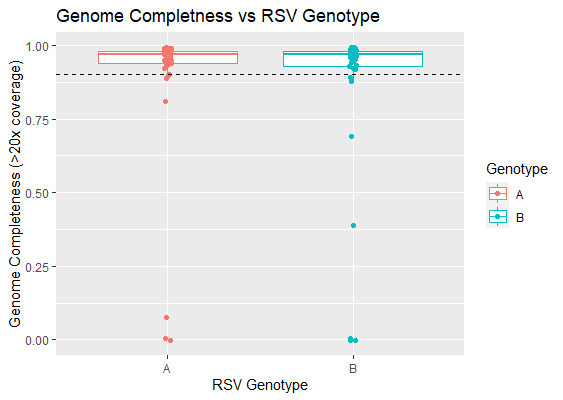
Figure 1: Genome Completeness vs RSV Genotype. Dashed line indicates 90% genome completeness.
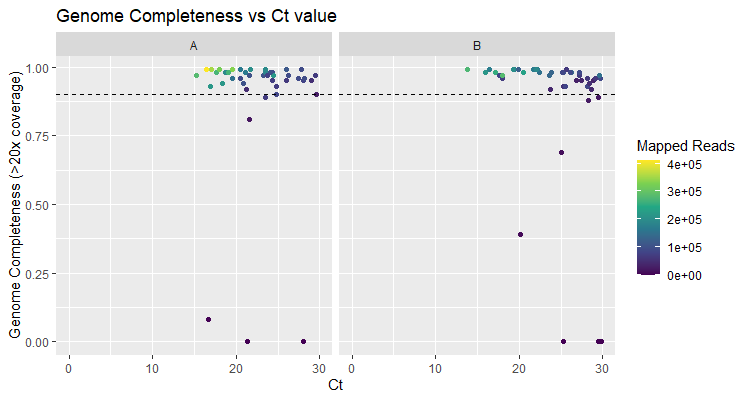
Figure 2: Genome Completeness vs Ct value. Dashed line indicates 90% genome completeness.
The samples included in these preliminary runs were a selection of RSV A and RSV B from the 2019/2020 and 2021/2022 epidemiological seasons. No significant difference in genome completeness was found when comparing these seasons for RSV A or B (Wilcoxon rank sum test, p > 0.05, n = 24 for 2019/2020 and 23 for 2021/2022 for both), suggesting these amplicon schemes performed at similar efficiencies regardless of sample age in our test set.
Using RAMPART to visualise coverage across the genome for each sample suggested some recurring amplicon dropouts, classified here as amplicons where the coverage did not reach the 20x depth threshold to be included in final consensus generation. For the RSV A primer scheme, the least efficient amplicon was amplicon 3, which amplified successfully in 24 (53.3%) out of 45 clinical samples. Overall, there was a median amplicon success rate of 41 samples (91.1%) succeeding per amplicon for the RSV A primer scheme. When using the RSV B primer scheme, amplicon 45 failed to reach the 20x depth threshold for any sample, the only amplicon that failed to amplify successfully, but with each amplicon responsible for only ~2% of genome coverage, or ~300 nucleotide bases on average, the loss of coverage was minimal. Overall amplicon success rate remained high, with the median amplicon succeeding in 40 (88.9%) out of 45 samples tested.
Initial phylogenetic analysis of the resulting RSV A and B genomes with a background set of high coverage genomes (i.e. full length genomes with >95% completeness) available on GISAID (n = 2465 RSV A and n = 2313 RSV B sequences; acknowledgement tables attached) showed that the samples sequenced in this study captured a range of diversity present in the tree, in particular for RSV A, suggesting that these primer schemes will be capable of amplifying a broad range of RSV genomes (Figure 3).
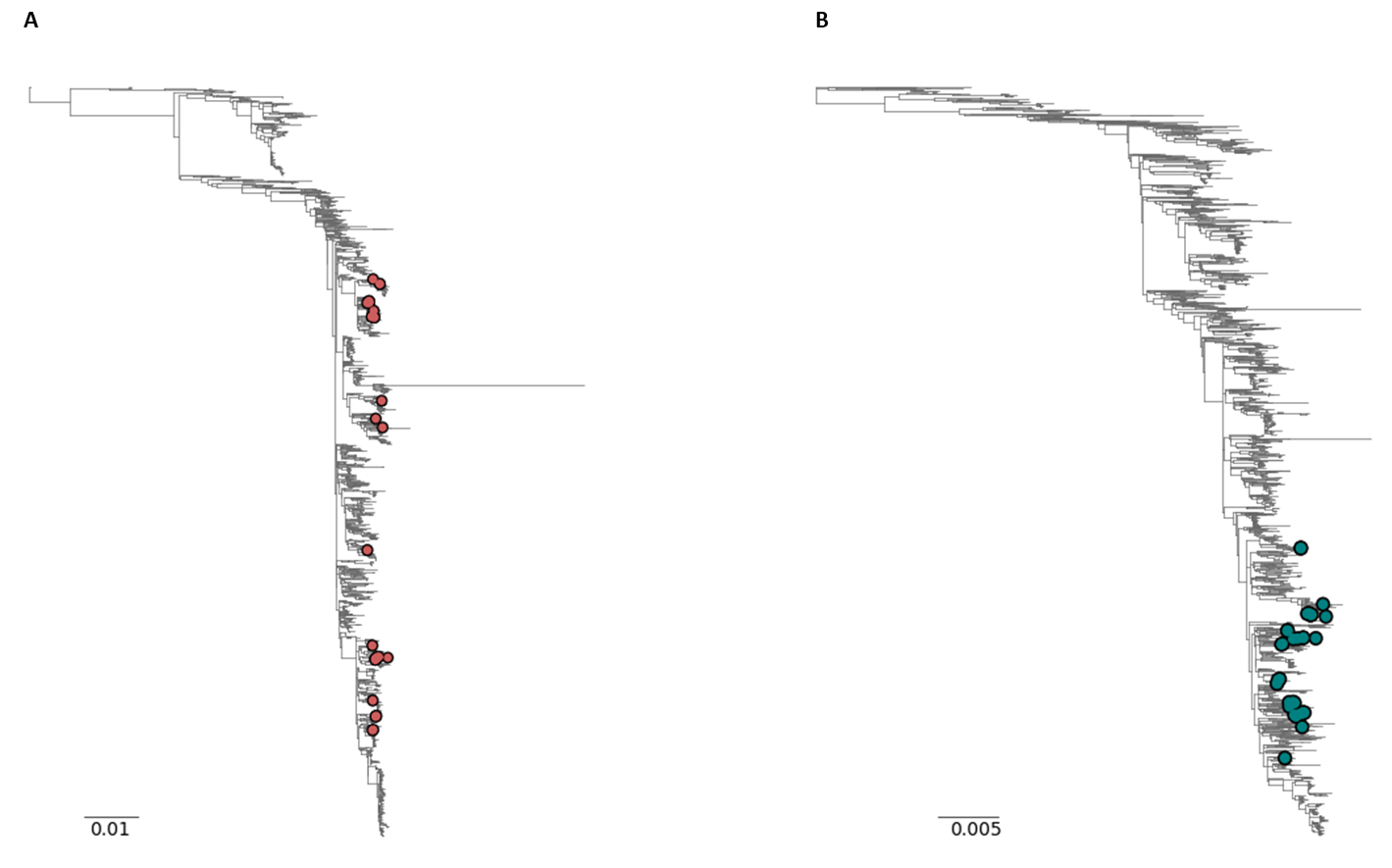
Figure 3: Phylogenetic tree of A) the 37 RSV A genomes (red circles) and B) the 36 RSV B genomes (blue circles) produced in this study with >90% genome completeness. Scale bar: substitutions per site
Discussion:
This preliminary testing of amplicon-based primer schemes for RSV A and RSV B has proved successful at harnessing existing SARS-CoV-2 sequencing infrastructure and has shown significant promise for allowing large-scale high-quality RSV genome production.
Work will continue to fully characterise these primer schemes, and to make them as efficient as possible. In particular, future work will increase the Ct cut off to find an upper limit for successful sequencing, the primer schemes will be tested on Illumina sequencing platforms, and efforts will be made to identify primers that can be spiked in to combat amplicon dropouts. In order to achieve this fully, a broader sample of RSV genomes needs to be sequenced, in particular as our current sample selection is geographically limited. As such, all sequences produced will be uploaded to NCBI and GISAID (post will be updated with accessions), and we will provide this RSV primer scheme open-source to enable other teams to benefit from this scheme.
Furthermore, we would love to hear from any teams globally that would like to collaborate to further test these primer schemes to their full potential.
References:
Templeton, K. E., Scheltinga, S. A., Beersma, M. F., Kroes, A. C., & Claas, E. C. (2004). Rapid and sensitive method using multiplex real-time PCR for diagnosis of infections by influenza a and influenza B viruses, respiratory syncytial virus, and parainfluenza viruses 1, 2, 3, and 4. Journal of clinical microbiology, 42(4), 1564–1569. https://doi.org/10.1128/JCM.42.4.1564-1569.2004
Quick, J., Grubaugh, N. D., Pullan, S. T., Claro, I. M., Smith, A. D., Gangavarapu, K., Oliveira, G., Robles-Sikisaka, R., Rogers, T. F., Beutler, N. A., Burton, D. R., Lewis-Ximenez, L. L., de Jesus, J. G., Giovanetti, M., Hill, S. C., Black, A., Bedford, T., Carroll, M. W., Nunes, M., Alcantara, L. C., Jr, … Loman, N. J. (2017). Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nature protocols, 12(6), 1261–1276. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples | Nature Protocols
Katoh, K., Misawa, K., Kuma, K., & Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic acids research, 30(14), 3059–3066. https://doi.org/10.1093/nar/gkf436
Jukes, T. H. and Cantor, C. R. (1969). Evolution of protein molecules in H. N. Munro, ed. Mammalian protein metabolism. Academic Press, New York. 21–132
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., & Lanfear, R. (2020). IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Molecular biology and evolution, 37(5), 1530–1534. https://doi.org/10.1093/molbev/msaa015
Acknowledgements
gisaid_RSV_acknowledgement_tables.zip (105.4 KB)