Preliminary Genomic Analysis of the RSV Surge 2022 in Massachusetts Reveals a Polyphyletic Epidemic Driven by the Expansion of Multiple Lineages
Introduction
As reported by the United States (US) Center for Disease Prevention and Control, the US experienced an early and severe respiratory syncytial virus (RSV) surge in autumn 2022 (Figure 1). This increase in RSV cases relative to previous years, alongside coincident epidemics of SARS-CoV-2 and Influenza, has put intense pressure on hospitals and urgent care centers [1]; however, the factors promoting the surge in cases are unknown. A leading hypothesis suggests an increase in the susceptible population brought on by limited RSV circulation during COVID-19, particularly involving children ages one to three, who are among the most susceptible to severe RSV [2]. Alternatively, the emergence of a highly transmissible and/or virulent “variant” of RSV could account for the increase. These possibilities can be distinguished by genome sequencing of surge-associated samples because a highly-transmissible variant will appear as a cluster of closely-related sequences in a phylogenetic tree. To characterize the viral genomic basis of the 2022 RSV surge in Massachusetts (MA), we sequenced RSV genomes and analyzed clinical data from symptomatic patients who presented to the Massachusetts General Hospital (MGH), a large academic medical center in Boston, MA, and its affiliated outpatient practices in the Greater Boston Area.
Results
Metagenomic sequencing of 108 samples produced 63 partial or complete genomes for downstream analysis
From early September to mid November 2022, MGH reported a surge in RSV cases, with a total of 950 RSV-positive samples (Table 1A). In the MGH dataset, the number of positive RSV cases peaked the week of October 29, 2022, much earlier than previous years, and subsequently fell, mirroring trends that occurred at the state and national level (Figure 1) [3]. This anomaly in timing and intensity of RSV circulation in 2022 has been noted on the global scale [4].
To characterize the RSV population underlying the surge, we performed unbiased whole-genome sequencing of 108 residual diagnostic upper respiratory tract specimens collected in November 2022. This resulted in 43 near-complete (> 80% genome covered) and 20 partial (>5% genome covered) genomes. We determined the RSV subtype for all 63 genomes, but only included near-complete genomes in subsequent phylogenetic analyses. Of the 108 samples for which sequencing was attempted, 84% (41/49) with a diagnostic qPCR cycle threshold (Ct) value ≤ 30 resulted in a genome, whereas only 3% (2/59) with a Ct > 30 resulted in a genome (Fisher’s exact test, p < 0.001) (Figure 2).
The sequenced cohort predominantly consists of symptomatic toddlers in acute care settings
The 63 patients from whom near-complete and partial genomes were assembled (“sequenced cases”) consisted of 44% (n = 28) males with a median age of 1 year, and with 19% (n = 12) collected in inpatient care, 40% (n = 25) collected in outpatient care, and 41% (n = 26) collected in the emergency room (Table 1B). All 63 patients reported symptoms consistent with a respiratory infection (Table 1B). Sequenced cases were representative of the 950 patients in the greater MGH RSV-positive patient pool with respect to multiple demographic criteria, including sex (p = 0.19, Fisher’s exact test), age (p = 0.07, Wilcoxon rank sum test), presenting hospital unit (p = 0.31, Fisher’s exact test), and presence of symptoms (p = 1, Fisher’s exact test). (Table 1A and B). This cohort also reflects the MGH outpatient testing algorithm, where the the Xpert Xpress SARS-CoV-2/Flu/RSV (Cepheid, Sunnyvale, CA) panel was recommended solely for symptomatic children under 3, immunocompromised patients, and individuals with chronic lung disease.
Diverse RSV genomes identified in the in the greater Boston area, with RSV-A predominating
Genomic analysis of the sequenced cases revealed that the surge was made up of by RSV-A: 90% (57/63 total; 40/43 near-complete) genomes were RSV-A while the remaining 10% (6/63 total; 3/43 near-complete) were RSV-B. We identified two instances of co-infection of RSV-A with Rhinovirus/Enterovirus. We did not detect the presence of other common respiratory viruses (Figure 3). We identified a single pair of near-complete genomes identical at the consensus-level, suggestive of a transmission link. Otherwise, genomic distances between samples ranged from 2 to 271 mutations for RSV-A, and 8 to 70 mutations for RSV-B, with a mean pairwise distance of 174 and 47 mutations respectively.
Phylogenetic inference reveals polyphyletic transmission of RSV-A
Phylogenetic comparison of the 40 near-complete RSV-A genomes suggests that the surge was not driven by a single lineage (Figure 4A); rather, the RSV-A surge thus far consists of at least 9 distinct clades, all with time to most recent common ancestors (tMRCAs) dated between 2014 and 2017 (Figure 4C). The tMRCA of all genomes in the dataset was at least 12 years ago, implying that the multiple RSV-A clades now circulating in MA diverged well before the COVID-19 pandemic, persisted throughout the COVID-19 pandemic, and have now expanded in association with the surge in cases. Without genomic surveillance data from more locations and intermediate time points (e.g. 2020 - 2021), it is not possible to resolve whether lineages expanded after a period of low-level/cryptic local transmission or were re-imported from external sites. Phylogenetic analysis of the 3 complete RSV-B genomes similarly yielded a tMRCA estimate of at least 6 years ago, in 2016 (Figure 4B, 4D). Like RSV-A, this clade likely circulated undetected prior to the autumn surge.
RSV-A and RSV-B genomes do not harbor higher than expected diversity
Highly transmissible viral variants may emerge that fundamentally alter epidemic dynamics. Surges caused by SARS-CoV-2 Alpha, Delta, and Omicron variants [5–7] have been linked to variants with a jump in genetic distance from the background epidemic. We found that the RSV-A genomes in our sample did not differ statistically from our estimated clock rate (10.8 substitutions per year), consistent with the hypothesis that the autumn 2022 surge was largely driven by typical lineages that have been circulating prior to SARS-CoV-2. Similarly, the RSV-B genomes harbor the expected amount of diversity in the context of the clock rate (12.4 substitutions per year) derived from the larger phylogenetic tree.
Discussion
We show that the expansion of multiple clades has contributed to the RSV surge in MA in the autumn of 2022. In cases sampled during the peak of the surge at a major academic medical center and its affiliated outpatient practices in the greater Boston area, RSV-A predominates, and circulating clades have a MRCA more than 5 years before the present. The polyphyletic nature of viral genomes sequenced in eastern MA suggests that the emergence of a single, highly transmissible RSV lineage is unlikely to account for the 2022 surge. Additional genomic and immunological data are needed to fully understand the causes of the 2022 RSV surge, including pre-COVID-19 pandemic sequences to evaluate changes in RSV genomic diversity during the COVID-19 pandemic.
Methods
Sample collection
We obtained 108 frozen (-80°C), archived upper respiratory tract samples that were positive for RSV from the MGH Clinical Microbiology Laboratory. Samples were from symptomatic individuals presenting for clinical care at MGH and its affiliated outpatient practices. Samples were tested for the presence of viral respiratory pathogens by one of three clinically verified, Food and Drug Administration (FDA) emergency use authorized or approved assays: the Xpert Xpress SARS-CoV-2/Flu/RSV (Cepheid, Sunnyvale, CA); the Xpert Xpress SARS-CoV-2/Flu/RSV Plus (Cepheid, Sunnyvale, CA); or the BioFire Respiratory 2.1 Panel (BioFire Diagnostics, Salt Lake City, UT). Samples were deidentified, and clinical data was obtained in accordance with the IRB (MGB-IRB 2019P003305).
RSV quantification and sequencing
To prepare samples for sequencing, we extracted total nucleic acid from upper respiratory tract samples in transport media, removed DNA with DNase I treatment, and assessed viral quantity using an RSV specific SYBR green RT-qPCR assay [8]. Human ribosomal RNA was then depleted with an RNase-H based method, and libraries were constructed using strand-specific ligation-based method [9]. Briefly, RNA was heat-fragmented and first-strand cDNA was generated with randomly primed reverse transcription. Second-strand cDNA was generated with nick translation and labeled with dUTP. Full-length, y-shaped, unique-dual-index Illumina sequencing adapters containing a UMI adjacent to the i7 index were ligated to the resulting double-stranded DNA. Samples were then treated with Uracil-Specific Excision Reagent (USER enzyme) to excise dUTP from the second strand. Libraries were PCR amplified, quantified, and pooled in equimolar ratios for sequencing on Illumina MiSeq or NextSeq 550 instruments.
RSV genome assembly and analysis
To assemble RSV genomes, we conducted all analyses using viral-ngs 2.1.28 10 on the Terra platform (app.terra.bio). All of the workflows named below are publicly available via the Dockstore Tool Registry Service (dockstore.org/organizations/BroadInstitute/collections/pgs). Briefly, samples were demultiplexed, reads were filtered for known sequencing contaminants, and de novo assembly with scaffolding against RSV-A (GenBank: KY654518.1) and RSV-B (GenBank: MZ516105.1) was performed.
The mean unambiguous genome length of these genomes was 12,371 bp for RSV-A and 9,960 bases for RSV-B. Assembled genomes with ≥ 50% completeness were deposited into NCBI GenBank. Raw reads for all samples (including those that did not produce a successful genome) were deposited in NCBI SRA. All NCBI data were deposited under BioProject: PRJNA904288.
Phylogenetic analysis
We constructed RSV-A and RSV-B specific maximum-likelihood (ML) phylogenetic trees [11] with associated visualizations using an augur pipeline [12] (augur_from_assemblies), part of the Nextstrain project [13]. We included all contextual genomes from GenBank with reported collection dates and a genome length greater than 12,160, corresponding to 80% of the reference genome length (resulting in a total of RSV-A N=1,228; RSV-B N=933 genomes on Dec 6, 2022). Within augur, RSV-A and RSV-B genomes were aligned via MAFFT v.7 to GenBank NC_038235.1 and NC_001781.1, respectively. This alignment was further processed in the ML analysis.
We used the contextualized ML phylogeny to calculate the number of clades circulating and estimate the time to tMRCA. To do so, we assigned a binary trait to each genome in the phylogeny, associated with the genome division of collection, and used Nextstrain’s ancestral inference to infer the state of that trait for each internal node in the tree. We defined a clade at the first node attributed to contain only MA descendants. Using baltic [14], we extracted these changes from the phylogenetic tree and plotted the inferred tMRCA for each clade using matplotlib [15].
Using the molecular clock rate regression line calculated by augur, we determined the residuals (number of mutations) per sample. Samples with a residual falling outside of the 2.5 – 97.5th percentiles were considered to statistically deviate from the molecular clock rate.
Coinfection analysis
All samples underwent multiplex PCR clinical testing with either a Flu-COVID-PCR multiplex assay or the Biofire Respiratory Virus panel. In the samples that underwent genomic sequencing and resulted in complete genomes, no co-infections were detected by clinical testing.
To assess for viral coinfections in unbiased sequencing, taxonomic classification of reads was performed with Kraken2 [16] using a custom database as previously described [17]. Viral taxa were considered present if >100 reads were classified to that taxa and filtered to include only known human respiratory viruses. All Kraken2 classifications were verified by megablast on de novo contigs. A classification was considered true if the least common ancestor of the top e-value megablast hits agreed with the Kraken2 classification for at least one contig (≥ 200 bp).
Figures
A
B
Figure 1: A. Epidemiological curve of RSV cases at MGH, in MA, and nationally shows samples taken from peak surge. The number of PCR positive tests for RSV as reported by the CDC in MA (blue, left axis) and the US (slate gray, right axis); along with the number of RSV positive tests conducted at MGH (red, left axis). The 108 samples selected for sequencing were drawn from a Nov 2 - Nov 14 window. B. RSV hospitalization rates for the overall network for CDC’s RSV-NET for 2017 - 2022 are shown in shaded gray. MGH RSV cases for the same time period are plotted in red.
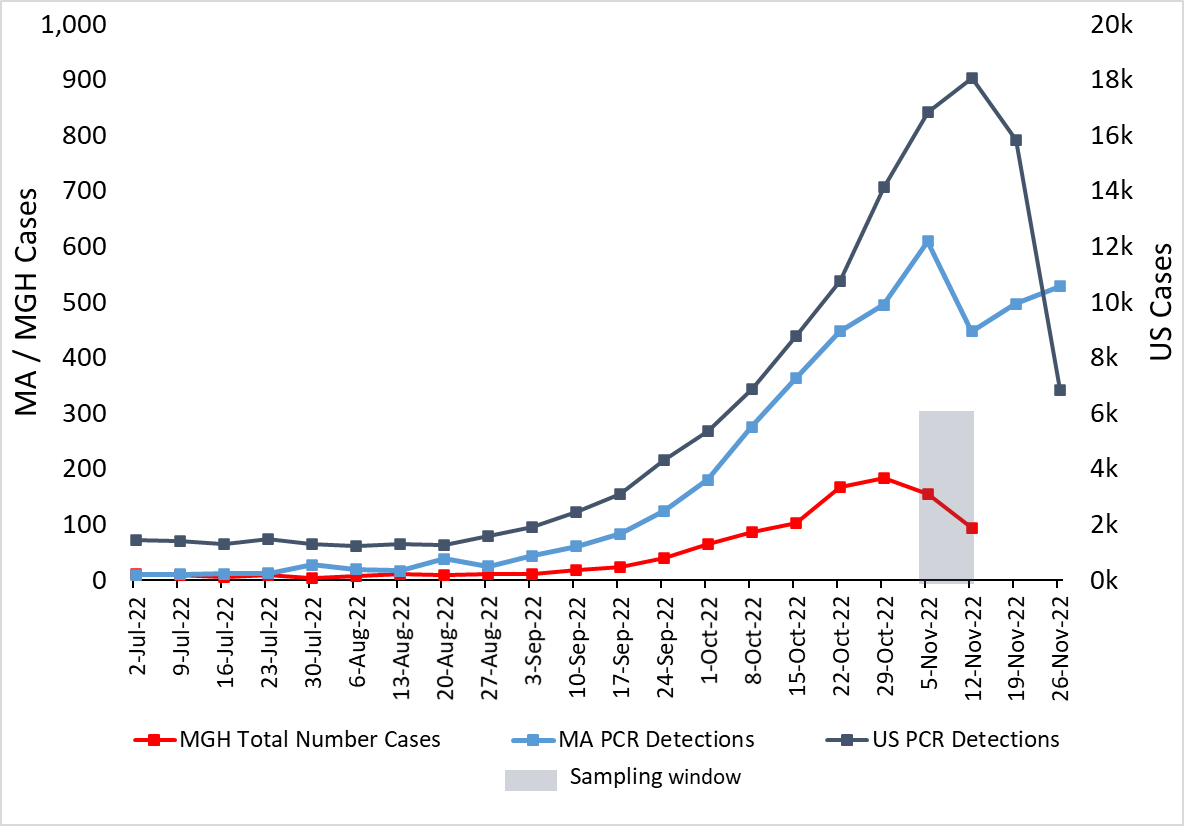
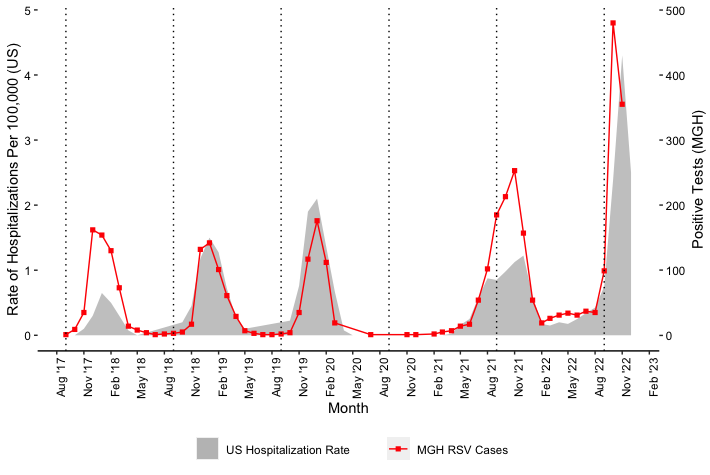
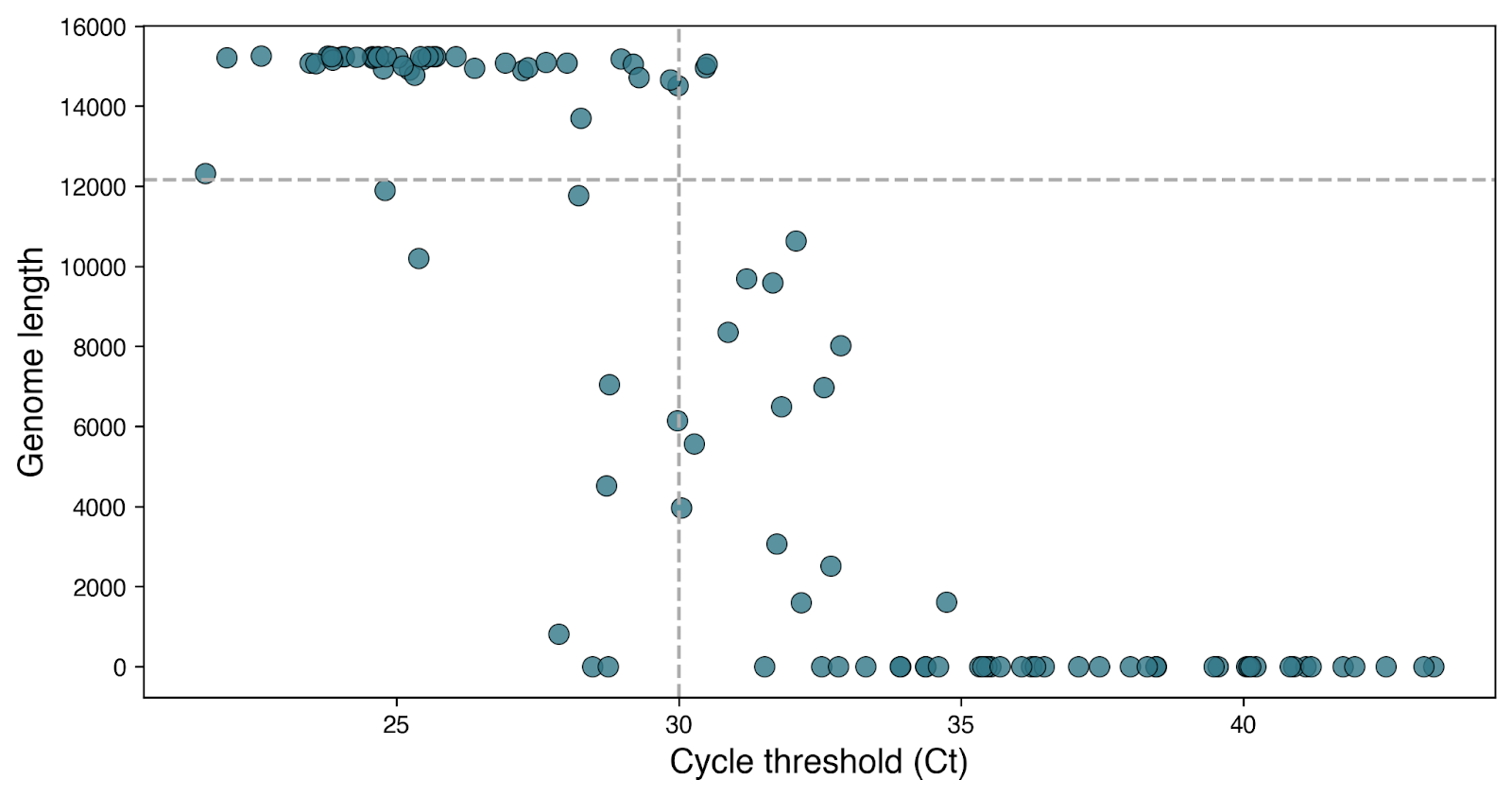
Figure 2: Unambiguous genome length vs cycle threshold (Ct). Each dot represents a unique sample in our dataset. Dashed lines are at a Ct of 30, and an unambiguous genome length of 12160, corresponding to ~80% of the RSV genome.
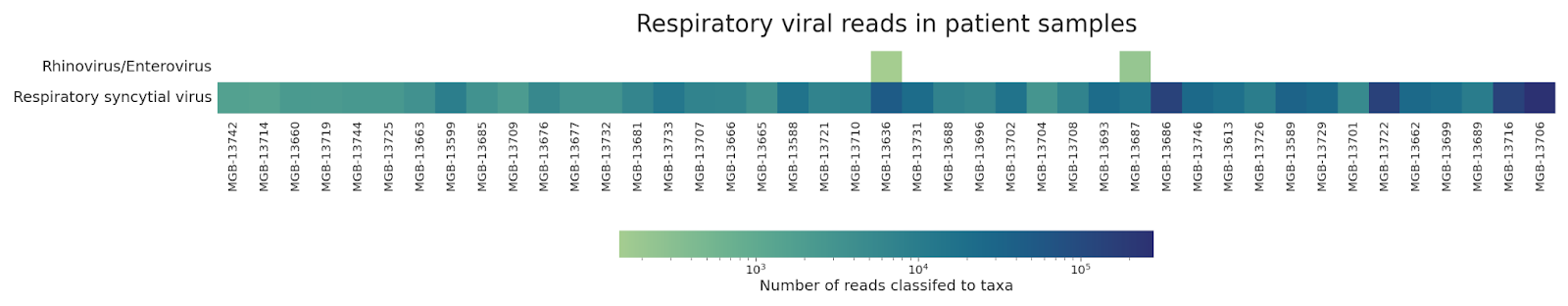
Figure 3: Coinfection analysis of unbiased sequencing reads. Taxonomic classification of reads from samples with near-complete RSV genomes by Kraken2. Shade represents the number of reads classified to a given taxa (log scale). Filtered to show known respiratory viral pathogens and only samples with >100 reads classified as a given viral taxa. Samples are sorted by RSV unambiguous assembly length from shortest (left) to longest (right).
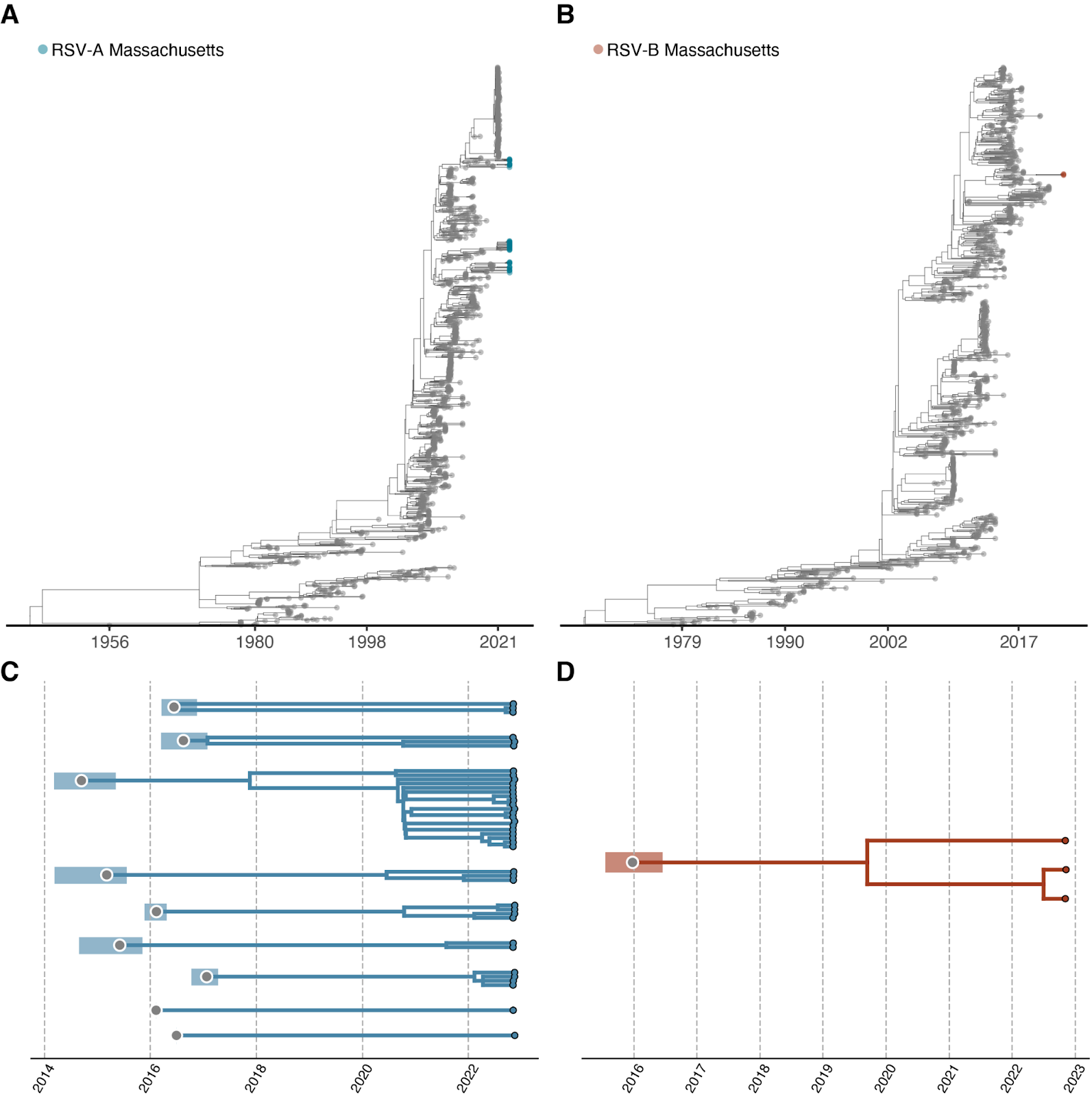
Figure 4: Phylogenetic Analysis of the outbreak. A) Maximum likelihood tree of all RSV-A genomes (N=1,139). Tips are colored according to division (MA tips in blue, all others in gray). B) Maximum likelihood tree of all RSV-B genomes (N=901). Tips are colored according to division (Massachusetts tips in orange, all others in gray). Explosion plot of C) RSV-A and D) RSV-B clades circulating in autumn 2022 in Eastern MA. Gray dots represent the inferred TMRCA for each clade. Shaded regions represent the confidence intervals associated with each tMRCA.

Table 1A. MGH Sample Demographics.
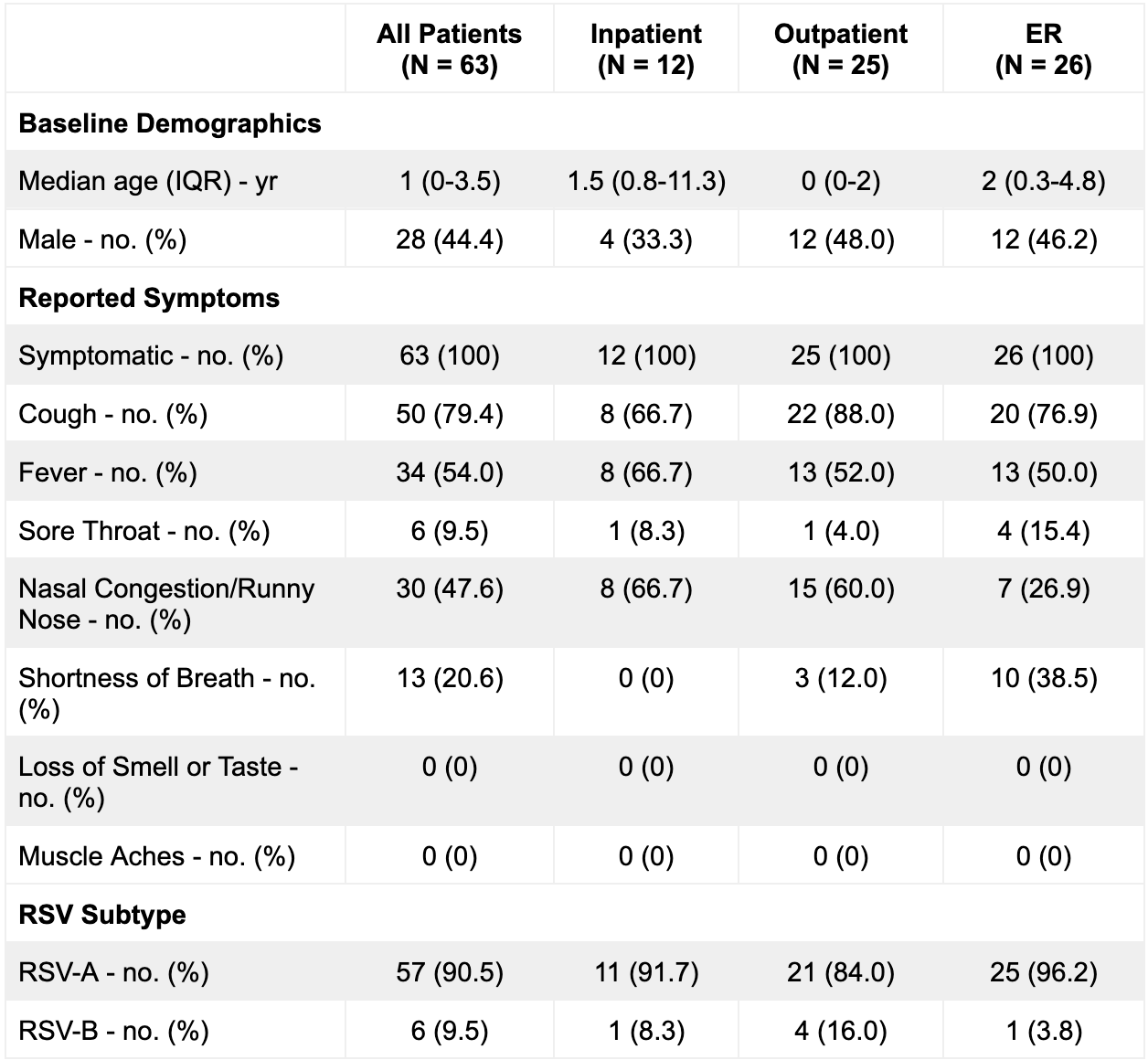
Table 1B. Sequenced Cohort Demographics.
Authors
RSV Genomic Analysis team includes:
Gordon Adams (1,2), Taylor Brock-Fisher (2), Sushma Chaluvadi (2), Katherine C. DeRuff (2), Sabrina Dobbins (2), Sanjat Kanjilal (7), Ben Kotzen (1,2), Jacob Lemieux (1,2), Zoe Levine (2,3,4), Christine Loreth (2), Bronwyn MacInnis (2), Katelyn Messer (2), Kristin Moffitt (8), Gage K. Moreno (2), Al Ozonoff (2,6), Daniel Park (2), Brittany A. Petros (2,3,9,10), Nira Pollock (8), Pardis Sabeti (2,11,12,13), Stephen F. Schaffner (2), Katie Siddle (2), Sarah Turbett (1), Rockib Uddin (1,2)
1 - Massachusetts General Hospital, Boston, MA, 02142.
2 - Broad Institute of MIT and Harvard, Cambridge, MA, 02142, USA.
3 - Harvard/Massachusetts Institute of Technology, MD-PhD Program, Boston, MA, USA.
4 - Harvard Program in Biological and Biomedical Sciences, Harvard Medical School, Boston, MA 02115, USA.
6 - Harvard Medical School, Boston, MA, 02115, USA.
7 - Brigham and Women’s Hospital, Boston MA. Boston, MA 02115.
8 - Boston Children’s Hospital, Boston, MA 02115.
9 - Division of Health Sciences and Technology, Harvard Medical School and Massachusetts Institute of Technology, Cambridge, MA, USA.
10 - Department of Systems Biology, Harvard Medical School, Boston, MA, USA.
11 - Harvard T.H. Chan School of Public Health, Boston, MA, 02115, USA.
12 - Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, MA, 02138, USA.
13 - Howard Hughes Medical Institute, Chevy Chase, MD, 20815, USA.
Contact:
[email protected] @bronwynmacinnis
[email protected] @dannyjpark
[email protected] @kjsiddle
[email protected] @sabeti_lab
[email protected]
Acknowledgements
This work was sponsored in part by the Centers for Disease Control Broad Agency Announcement (75D30120C09605 to B.L.M.), the Howard Hughes Medical Institute Investigator award to P.C.S., the National Institute of Allergy and Infectious Diseases (U19AI110818 to P.C.S), the Massachusetts Consortium on Pathogen Readiness (J.E.L), and the National Institute of General Medical Sciences (T32GM007753 to B.A.P.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Use of data
Raw reads and assembled genomes are submitted to genbank under PRJNA904288. The data are available immediately and shared under Genbank’s use agreements to facilitate accelerated public health and scientific investigations. A publication on these sequences from our group is forthcoming. Investigators who wish to conduct primary analysis of the sequence data in this report are asked to contact the authors to coordinate.
Conflicts of interest
Dr. Sabeti is a co-founder and consultant at Sherlock Biosciences Inc. and Delve Bio, and is a Board Member of Danaher Corporation; she holds equity in all three companies. She has several patents related to diagnostics, genome sequencing, and informatics, including two patents licensed to Sherlock Biosciences.
Ethical statement
This study was conducted at the Broad Institute and Massachusetts General with approval from the Massachusetts General Brigham Institutional Review Board under Protocol #2019P003305 and from the MIT Institutional Review Board under Protocol #1612793224.
References
- HAN Archive - 00479. 2022 [cited 2022 Dec 7].
- Billard MN, Bont LJ. Quantifying the RSV immunity debt following COVID-19: a public health matter. Lancet Infect Dis [Internet]. 2022 Sep 2
- RSV-NET interactive dashboard. 2022 [cited 2022 Dec 8].
- Eden JS, Sikazwe C, Xie R, Deng YM, Sullivan SG, Michie A, Levy A, Cutmore E, Blyth CC, Britton PN, Crawford N, Dong X, Dwyer DE, Edwards KM, Horsburgh BA, Foley D, Kennedy K, Minney-Smith C, Speers D, Tulloch RL, Holmes EC, Dhanasekaran V, Smith DW, Kok J, Barr IG, Australian RSV study group. Off-season RSV epidemics in Australia after easing of COVID-19 restrictions. Nat Commun. 2022 May 24;13(1):2884. PMCID: PMC9130497
- Vöhringer HS, Sanderson T, Sinnott M, De Maio N, Nguyen T, Goater R, Schwach F, Harrison I, Hellewell J, Ariani CV, Gonçalves S, Jackson DK, Johnston I, Jung AW, Saint C, Sillitoe J, Suciu M, Goldman N, Panovska-Griffiths J, Wellcome Sanger Institute COVID-19 Surveillance Team, COVID-19 Genomics UK (COG-UK) Consortium*, Birney E, Volz E, Funk S, Kwiatkowski D, Chand M, Martincorena I, Barrett JC, Gerstung M. Genomic reconstruction of the SARS-CoV-2 epidemic in England. Nature. 2021 Dec;600(7889):506–511. PMCID: PMC8674138
- [Mlcochova P, Kemp S, Dhar MS, Papa G, Meng B, Ferreira IATM, Datir R, Collier DA, Albecka A, Singh S, Pandey R, Brown J, Zhou J, Goonawardane N, Mishra S, Whittaker C, Mellan T, Marwal R, Datta M, Sengupta S, Ponnusamy K, Radhakrishnan VS, Abdullahi A, Charles O, Chattopadhyay P, Devi P, Caputo D, Peacock T, Wattal DC, Goel N, Satwik A, Vaishya R, Agarwal M, Indian SARS-CoV-2 Genomics Consortium (INSACOG), Genotype to Phenotype Japan (G2P-Japan) Consortium, CITIID-NIHR BioResource COVID-19 Collaboration, Mavousian A, Lee JH, Bassi J, Silacci-Fegni C, Saliba C, Pinto D, Irie T, Yoshida I, Hamilton WL, Sato K, Bhatt S, Flaxman S, James LC, Corti D, Piccoli L, Barclay WS, Rakshit P, Agrawal A, Gupta RK. SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature. 2021 Sep 6;
- Viana R, Moyo S, Amoako DG, Tegally H, Scheepers C, Althaus CL, Anyaneji UJ, Bester PA, Boni MF, Chand M, Choga WT, Colquhoun R, Davids M, Deforche K, Doolabh D, du Plessis L, Engelbrecht S, Everatt J, Giandhari J, Giovanetti M, Hardie D, Hill V, Hsiao NY, Iranzadeh A, Ismail A, Joseph C, Joseph R, Koopile L, Kosakovsky Pond SL, Kraemer MUG, Kuate-Lere L, Laguda-Akingba O, Lesetedi-Mafoko O, Lessells RJ, Lockman S, Lucaci AG, Maharaj A, Mahlangu B, Maponga T, Mahlakwane K, Makatini Z, Marais G, Maruapula D, Masupu K, Matshaba M, Mayaphi S, Mbhele N, Mbulawa MB, Mendes A, Mlisana K, Mnguni A, Mohale T, Moir M, Moruisi K, Mosepele M, Motsatsi G, Motswaledi MS, Mphoyakgosi T, Msomi N, Mwangi PN, Naidoo Y, Ntuli N, Nyaga M, Olubayo L, Pillay S, Radibe B, Ramphal Y, Ramphal U, San JE, Scott L, Shapiro R, Singh L, Smith-Lawrence P, Stevens W, Strydom A, Subramoney K, Tebeila N, Tshiabuila D, Tsui J, van Wyk S, Weaver S, Wibmer CK, Wilkinson E, Wolter N, Zarebski AE, Zuze B, Goedhals D, Preiser W, Treurnicht F, Venter M, Williamson C, Pybus OG, Bhiman J, Glass A, Martin DP, Rambaut A, Gaseitsiwe S, von Gottberg A, de Oliveira T. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature. 2022 Mar;603(7902):679–686. PMID: 35042229
- Sanghavi SK, Bullotta A, Husain S, Rinaldo CR. Clinical evaluation of multiplex real-time PCR panels for rapid detection of respiratory viral infections. J Med Virol. 2012 Jan;84(1):162–169. PMCID: PMC7166524
- Matranga CB, Andersen KG, Winnicki S, Busby M, Gladden AD, Tewhey R, Stremlau M, Berlin A, Gire SK, England E, Moses LM, Mikkelsen TS, Odia I, Ehiane PE, Folarin O, Goba A, Kahn SH, Grant DS, Honko A, Hensley L, Happi C, Garry RF, Malboeuf CM, Birren BW, Gnirke A, Levin JZ, Sabeti PC. Enhanced methods for unbiased deep sequencing of Lassa and Ebola RNA viruses from clinical and biological samples. Genome Biol. 2014;15(11):519. PMCID: PMC4262991
- Park DJ, Dudas G, Wohl S, Goba A, Whitmer SLM, Andersen KG, Sealfon RS, Ladner JT, Kugelman JR, Matranga CB, Winnicki SM, Qu J, Gire SK, Gladden-Young A, Jalloh S, Nosamiefan D, Yozwiak NL, Moses LM, Jiang PP, Lin AE, Schaffner SF, Bird B, Towner J, Mamoh M, Gbakie M, Kanneh L, Kargbo D, Massally JLB, Kamara FK, Konuwa E, Sellu J, Jalloh AA, Mustapha I, Foday M, Yillah M, Erickson BR, Sealy T, Blau D, Paddock C, Brault A, Amman B, Basile J, Bearden S, Belser J, Bergeron E, Campbell S, Chakrabarti A, Dodd K, Flint M, Gibbons A, Goodman C, Klena J, McMullan L, Morgan L, Russell B, Salzer J, Sanchez A, Wang D, Jungreis I, Tomkins-Tinch C, Kislyuk A, Lin MF, Chapman S, MacInnis B, Matthews A, Bochicchio J, Hensley LE, Kuhn JH, Nusbaum C, Schieffelin JS, Birren BW, Forget M, Nichol ST, Palacios GF, Ndiaye D, Happi C, Gevao SM, Vandi MA, Kargbo B, Holmes EC, Bedford T, Gnirke A, Ströher U, Rambaut A, Garry RF, Sabeti PC. Ebola Virus Epidemiology, Transmission, and Evolution during Seven Months in Sierra Leone. Cell. 2015 Jun 18;161(7):1516–1526. PMCID: PMC4503805
- Sagulenko P, Puller V, Neher RA. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018 Jan;4(1):vex042. PMCID: PMC5758920
- Huddleston J, Hadfield J, Sibley TR, Lee J, Fay K, Ilcisin M, Harkins E, Bedford T, Neher RA, Hodcroft EB. Augur: a bioinformatics toolkit for phylogenetic analyses of human pathogens. J Open Source Softw Internet. 2021 Jan 7;6(57).
- Hadfield J, Megill C, Bell SM, Huddleston J, Potter B, Callender C, Sagulenko P, Bedford T, Neher RA. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 2018 Dec 1;34(23):4121–4123. PMCID: PMC6247931
- Dudas G. baltic: baltic - backronymed adaptable lightweight tree import code for molecular phylogeny manipulation, analysis and visualisation.
- Hunter. Matplotlib: A 2D Graphics Environment. 2007 May 1;9:90–95.
- Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019 Nov 28;20(1):257. PMCID: PMC6883579
- Lemieux JE, Siddle KJ, Shaw BM, Loreth C, Schaffner SF, Gladden-Young A, Adams G, Fink T, Tomkins-Tinch CH, Krasilnikova LA, DeRuff KC, Rudy M, Bauer MR, Lagerborg KA, Normandin E, Chapman SB, Reilly SK, Anahtar MN, Lin AE, Carter A, Myhrvold C, Kemball ME, Chaluvadi S, Cusick C, Flowers K, Neumann A, Cerrato F, Farhat M, Slater D, Harris JB, Branda JA, Hooper D, Gaeta JM, Baggett TP, O’Connell J, Gnirke A, Lieberman TD, Philippakis A, Burns M, Brown CM, Luban J, Ryan ET, Turbett SE, LaRocque RC, Hanage WP, Gallagher GR, Madoff LC, Smole S, Pierce VM, Rosenberg E, Sabeti PC, Park DJ, MacInnis BL. Phylogenetic analysis of SARS-CoV-2 in Boston highlights the impact of superspreading events. Science [Internet]. 2021 Feb 5;371(6529).