Introduction
The Ministry of Health, Uganda declared an outbreak of the Sudan Ebola Virus on 20th September 2022 after the virus was identified in an individual from Mubende district by the Uganda Virus Research Institute (UVRI) on the 19th of September 2022. (September 2022 Sudan Ebola Virus Disease Outbreak in Uganda - Ebolavirus, 2022). Subsequently, cases were identified in several other districts including, Kyegegwa, Kassanda, Kagadi, Wakiso, Kampala, Masaka, Jinja, and Bunyangabu through the months of September, October, and November.
Methods
In an effort to support and build capacity for pathogen genomic surveillance, the Central Public Health Laboratories, Uganda (CPHL) with support from Africa Centre for Disease Control and Institut National de Recherche Biomédicale (INRB), Kinshasa, Democratic Republic of Congo (DRC) sequenced randomly selected samples (n= 58) collected in the months of October and November from Mubende and Kassanda districts, the epicenters of the outbreak. Samples were inactivated in the field laboratory, nucleic acid was extracted, libraries prepared, and long-read cDNA sequenced with the MinION Mk1C using protocols adopted from the INRB at National Genomics Reference Laboratory (NGRL) CPHL. To validate the MiniION results, samples with qubit values more than 10ng (n=32) were sequenced on the illumina platform. Consensus genomes were generated using the ARTIC pipeline. Multiple sequence alignment of genomes with coverage more than 70% and Ct values less than 30 was performed using the web version of MAFFT v7.745 (MAFFT - a multiple sequence alignment program). (Katoh et al., 2019). The resulting file was subjected to SNP-sites v2.5.1 to generate a PHYLIP file. (Page et al., 2016). The file was used by PhyML to build a phylogenetic tree which was visualized by the Interactive Tree of Life (iTOL v6) and rooted using the Sudan ebolavirus reference (NC_006432.1). (Guindon et al., 2010; Letunic & Bork, 2007).
Variants were annotated from the ARTIC pipeline with snpEff v5.0.1 and using customized scripts, missense variants were extracted, and a sample-by-sample comparison performed.
Results and Discussion
From MinION results, 41 samples had genome coverage of 70% and above while 17 had coverage below 70%. Whereas on the Illumina platform, 30 samples had genome coverage of 70% and above while 2 had coverage below 70% as summarized in the table below.
| Coverage Classification | MinION Count | Illumina count |
|---|---|---|
| Above 70% | 41 (70.7%) | 30 (93.8%) |
| Below 70% | 17 (29.3%) | 2 (6.2%) |
| Total | 58 | 32 |
From the phylogenetic analysis (Fig. 1), the samples are closely related with the Nakisamata Sudan Ebolavirus strain that emerged in Luwero District, Uganda in May 2011. This is consistent with the earlier results of the genome sequenced by the UVRI (Mubende2022) that also clustered with our samples (colored in blue, Fig.1).

Figure 1: Phylogenetic tree showing the evolutionary relationship of the current (2022) outbreak and Nakisamata sample
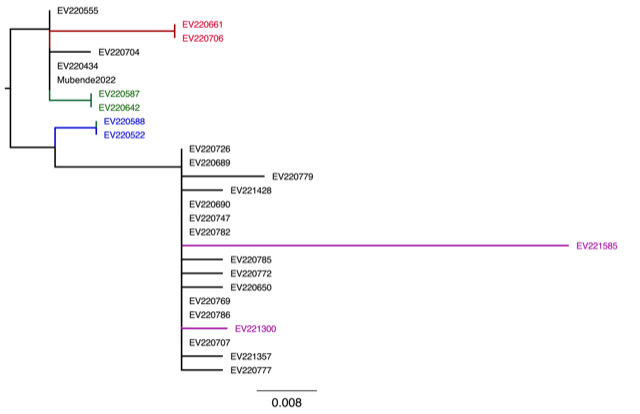
From the variant analysis, specimen EV220706 and EV220661 (Red Fig.2) collected on 24th October and 23rd October respectively from Mubende district exclusively shared a common single nucleotide polymorphism (SNP) mutation (c.6556G>A) which affects the second secreted glycoprotein and leads to a change from amino acid alanine to threonine at position 187 (p.Ala187Thr). Glycoprotein mutations were previously observed during the 2013-2016 Ebola virus outbreak in West Africa, and these were associated with increased viral infectivity in vitro (Fels et al., 2021.). This is an area for be further investigation for Ebola Sudan in our setting.
We also observed that EV220587 (Green Fig.2) collected on 22nd October 2022 and EV220522 (Blue Fig.2) collected on 20th October clustered very closely with their repeat samples EV220642 (Green Fig.2) and EV220588 (Blue Fig.2) collected 23rd and 22nd October respectively. However, we found that EV221300 (Pink Fig.2) collected on 04th November and its respective repeat sample EV221585 (Pink Fig.2) collected on the 10th of November 2022 did not cluster closely as observed for the other repeat sample pairs. On deeper investigation, we found that each of these specimens had unique mutations; c.15958T>G for EV221300 leading to a change in amino acid leucine to tryptophan at position 1475 (p.Leu1475Trp) and c.14971A>G for EV221585 leading to a change in amino acid lysine to arginine at position 1146 (p.Lys1146Arg) that were absent in the rest of the samples tested in this sequencing run. Both of these unique mutations are known to affect the same gene product - RNA-dependent RNA polymerase. A further investigation with more samples will be required to better understand the possible relevance of these mutations with respect to the current Ebola Sudan outbreak as well as future outbreaks in Uganda.
Data availability
Sequence data is available here (GitHub - Kanyerezi30/CPHL-EBOV)
Partners and collaborators
Susan Nabadda (NHLDS/CPHL/UNHLS, Uganda)
Isaac Ssewanyana (NHLDS/CPHL/UNHLS, Uganda)
Alisen Ayitewala (NHLDS/CPHL/UNHLS, Uganda)
Aloysious Ssemaganda (NHLDS/CPHL/UNHLS, Uganda,)
Stephen Kanyerezi (NHLDS/CPHL/UNHLS, Uganda)
Ivan Sserwadda (NHLDS/CPHL/UNHLS, Uganda)
Pimundu Godfrey (NHLDS/CPHL/UNHLS, Uganda)
Sofonias Tessema (Africa CDC, Ethiopia)
Gerald Mboowa (Africa CDC, Ethiopia)
Placide Mbala-Kingebeni (INRB, University of Kinshasa)
Dorcas Waruguru Wanjohi (Africa CDC, Ethiopia)
Jean-Jacques Muyembe (INRB, University of Kinshasa)
Adrienne Amuri-Aziza (INRB, University of Kinshasa)
Jean Claude Makangara (INRB, University of Kinshasa)
Gradi Luakanda (INRB, University of Kinshasa)
Andrew Rambaut (University of Edinburgh)
Kakooza Francis (Infectious Diseases Institute, Makerere University)
We gratefully acknowledge the Africa CDC’s Africa Pathogen Genomics Initiative (Africa PGI) for support for equipment and reagents.
Statement on continuing work and analyses prior to publication
These genomes are being shared pre-publication. Please note that this data is based on work in progress and should be considered preliminary. Our analysis of these data is ongoing and a publication communicating our findings is in preparation. If you intend to use these sequences prior to our publication, please communicate with Dr. Isaac Ssewanyana for coordination.