The recent outbreak aboard the MV Hondius caused by Andes virus raises two key questions [1]: (i) where do the outbreak genomes cluster geographically, and (ii) how can the phylogenetic signal be reconciled with the documented travel history and timing of symptoms onset in the index case.
To explore these questions, we compared the recently released complete genome from case 7 (a passenger who disembarked at St Helena on April 22) with rodent-derived genomes we recently generated from southern Chile and all publicly available Orthohantavirus andesense genomes with >75% coverage per segment. At the Programa Hantavirus y Zoonosis, Universidad del Desarrollo - Clínica Alemana (Chile), we’ve worked on a genomic surveillance framework for Andes virus (ANDV) based on a rapid amplicon-based ONT approach. As a pilot study, we generated near-complete ANDV genomes from rodents captured in peridomestic settings associated with suspected sites of human infection in southern Chile (accessed up to: https://papers.ssrn.com/sol3/papers.cfm?abstract_id=6620250). The dataset currently includes rodents sampled from four distinct localities (Fig. 1), with at least two rodents sequenced per site. We reconstructed maximum-likelihood phylogenies for the L, M and S segments independently using IQ-TREE2 under modelFinder with the ultrafast bootstrap method with 1,000 replicates.
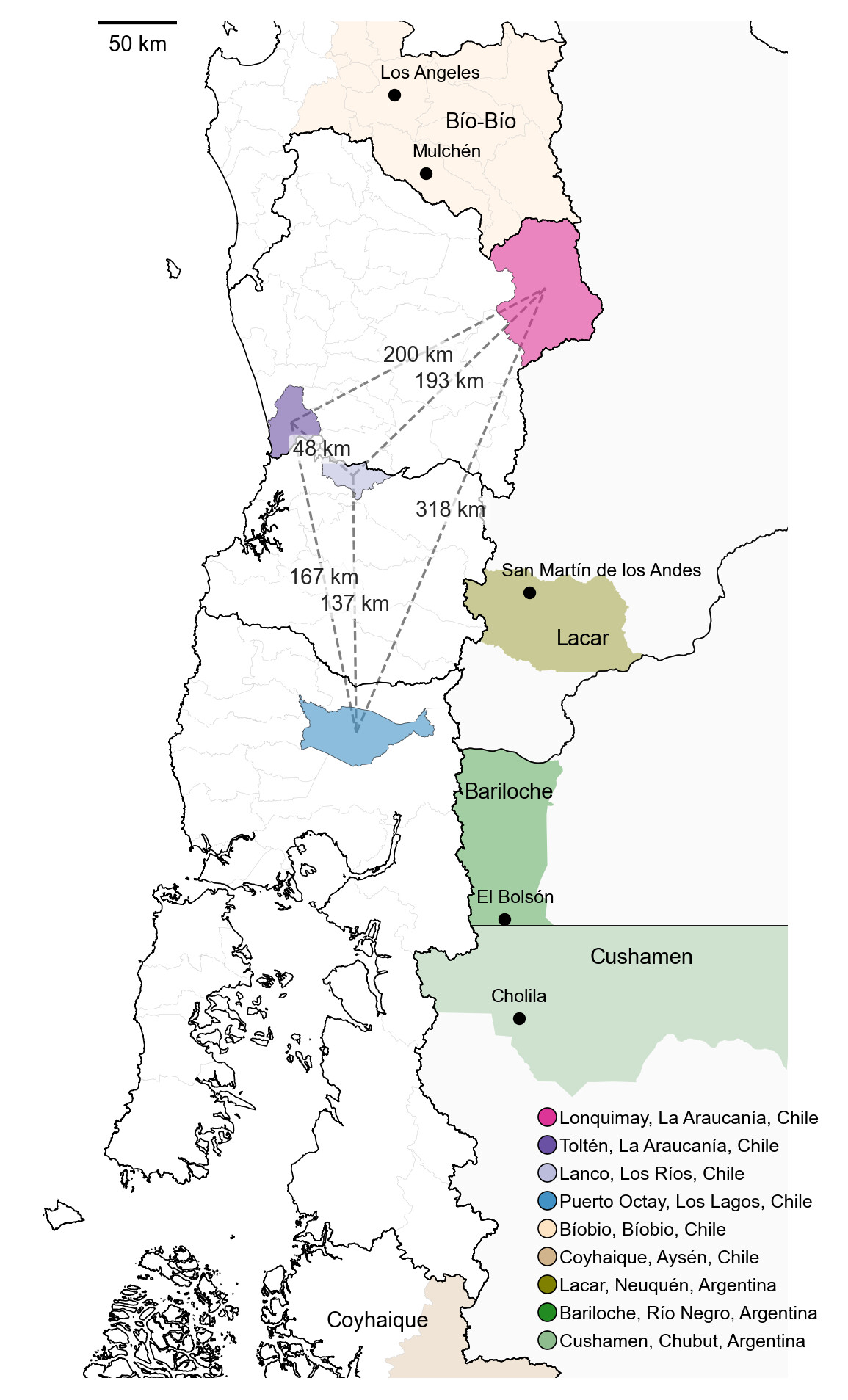
| Figure 1. Map showing the administrative regions of Bíobio, La Araucanía, Los Ríos, Los Lagos, Aysén in southern Chile and Neuquén, Río Negro and Chubut provinces in southern Argentina, respectively. Distances between localities where the selected rodents were captured are indicated in kilometers. |
Phylogenetically related viruses within the Orthohantavirus andense species, including Laguna Negra virus (LANV), Lechiguanas virus (LECV), Orán virus (ORV), Buenos Aires virus (BASV), circulate in north-central Argentina, Paraguay and southern Brazil, reflecting the broad geographic distribution of O. andesense throughout southern South America (Fig 2). Within the Andes virus lineage, genomes associated with documented Argentine transmission events – including the 2018-19 Epuyén super-spreader outbreak [2], the Río Negro household transmission cluster [3] and the Epilink/96 [4] sequence associated with the first known person-to-person case – formed a well-supported monophyletic clade (>88 bootstrap support) across all three segments (Fig. 3). However, this clade is not exclusively comprised of outbreak-associated viruses, as it also includes other genomes such as NRC-2/17 and NRC-6/18, which are derived from unrelated human cases from nearby endemic provinces. No rodent-derived genomes are currently represented within this clade.
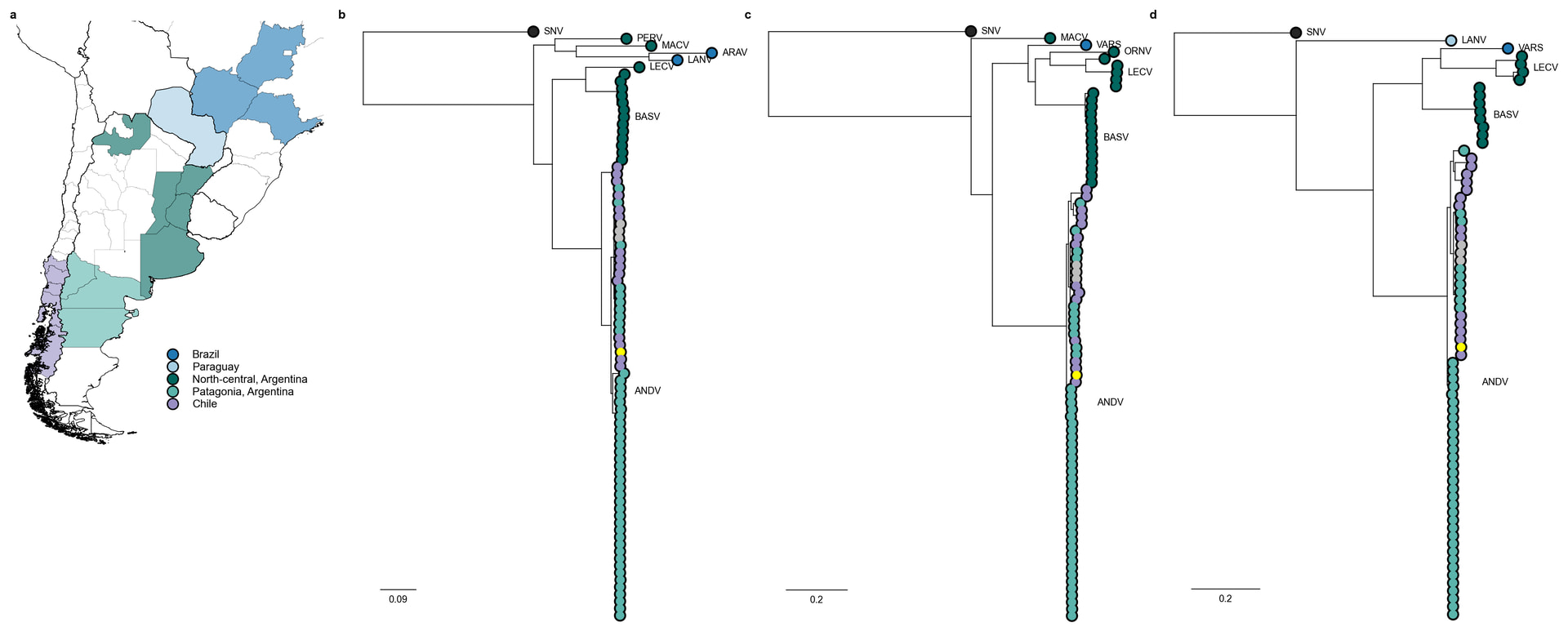
| Figure 2. Geographical distribution and phylogenetic analyses of Orthohantavirus andesense genome segments. (a) Geographic origin of O. andesense sequences included. (b-d) Segment-specific bayesian trees inferred for the S (b), M (c) and L (d) segments. Tips are colored according to macro-region of origin, consistent with panel (a). The outbreak-associated sequence is highlighted in yellow. |
Across all three segments, the MV Hondius outbreak-associated genome clusters most consistently within the Andes lineage circulating in southern Chile and adjacent Argentine provinces including Neuquén, Río Negro and Chubut (Fig. 1, 2a). The outbreak lineage showed the closest phylogenetic relationship to rodent-derived viruses sampled in southern Chile, particularly our new sequences R175 and R190 from Toltén in the La Araucanía region (Fig.1), as well as R577 and R589 from Lanco in the southern Los Ríos region, and NRC-3 and NRC-4 derived from human-cases in the bordering San Martín de los Andes (Neuquén, Argentina) with strong bootstrap support across segment phylogenies (>93; Fig. 3). The outbreak lineage is separated from sequences from the Chilean rodent reservoir sequences by branch lengths ranging between 0.004 and 0.008 substitutions/site across all segments, representing 8-11, 25-30 and 41-50 SNPs for the S, M and L segments respectively. Despite minor variation within the cluster - the outbreak lineage M segment clusters most closely with R175 and R190 in the L segment respectively - the overall congruence across segment phylogenies suggests the absence of detectable reassortment events in the outbreak lineage. NRC-3 and NRC-4 genomes were generated in the study describing the 2018 Epuyén super-spreader event in Argentina by Martinez et al. 2020, although both cases were classified as epidemiologically unrelated to the outbreak. This clustering pattern has been previously highlighted [5, 6].

| Figure 3. Maximum-likelihood phylogenies of the ANDV clade for the S, M and L segments. Tips are colored by sampling location. Numbers above branches indicate bootstrap support of selected nodes |
The Chilean sampling localities of Toltén and Lanco are approximately 48 km apart, and located more than 100 km west of the Argentine border, whereas San Martin de los Andes is ~150 km east of Toltén and Lanco (Fig.1). These genomes define a geographically localized trans-Andean clade spanning southern Chile and northwestern Argentine Patagonia. The Andes mountain range, an ecological and political border, likely plays an important role in structuring the population connectivity and dispersal dynamics of Oligoryzomys longicaudatus (long-tailed pygmy rice rat), the principal ANDV reservoir host. Although high elevations may represent a significant barrier for rodent populations, lower-altitude Andean corridors in southern Patagonia could facilitate trans-Andean dispersal and historical connectivity among host populations [7]. Southern populations of O. longicaudatus show high dispersal capacity and genetic homogeneity, although individual mobility remains spatially restricted at local scales (home range ~4.5km). The ANDV reservoir host is distributed from the southern Atacama desert (~27ºS) to Tierra del fuego (~54ºS), spanning major ecogeographic regions including Mediterranean, the Temperate, and the Patagonian Forests [8]. In this context, geographically localized lineages underscore unresolved questions regarding host population connectivity, the ecological drivers of host population structure and the maintenance of ANDV across southern Andean regions.
Table 1. Possible scenarios for the zoonotic origin of the outbreak lineage.
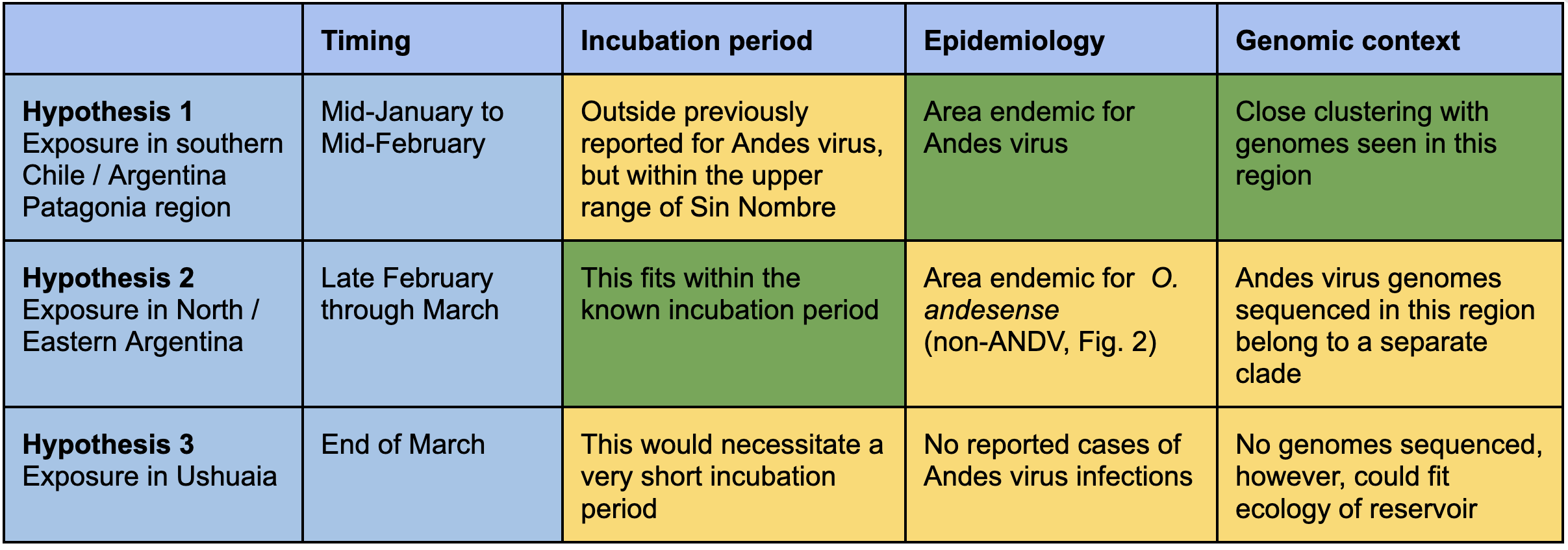
Travel records indicate that the index case traveled through southern Chile and Argentina (La Araucanía, Los Ríos and Neuquén) during late January through February 20, 2026 (Fig. 4). The case then boarded the MV Hondius cruise at Ushuaia on March 30. Symptom onset was reported on April 6 and death on April 11. Based on currently available genomic and epidemiological data, several hypotheses emerge (Table 1): (1) Exposure in southern Chile-Argentina region during late January through February, would explain the close phylogenetic clustering. However, this scenario would imply a very long incubation period. Depending on the timing of exposure, this scenario would suggest an incubation period ranging from ~45-76 days before symptoms onset on April 6, which is at the upper range reported for environmental acquired ANDV which may extend up to ~40 days [9, 10]. (2) Alternatively, the Toltén/Lanco/Neuquén-associated trans-Andean clade circulates more broadly across southern South America than currently observed given the limited genomic representation. In this scenario, the timeline would fit better with the expected incubation period, as exposure could have occurred late during travel through northern Argentina (February 24 to March 11). However no currently available genomes from these regions cluster closely with the outbreak lineage, and given the strong geographic structure across the O. andesense phylogenies discussed above, such a widespread distribution of this clade appears ecologically unlikely. Northern Argentina and the southern Andean regions are separated by substantial geographic distance, major ecological transitions and marked differences in Andean topography including substantial changes in elevation (Fig. 2). (3) A third possibility would imply exposure in Ushuaia, Argentina before boarding the MV Hondius cruise (Fig. 4). This scenario would better fit a shorter incubation period, but it would additionally imply ANDV circulation in the archipelago Tierra del Fuego. Although the reservoir host population extends as far south as Tierra del Fuego, all currently available ANDV sequences originate from localities along the Andean regions of southern Chile and Argentina. Despite this broad distribution, no hantavirus cases have been reported in the southernmost region of Chile (Magallanes y la Antártica Chilena) or in Ushuaia in Tierra del Fuego – the departure point of the MV Hondius cruise ship – which is located approximately 1,500 km southeast from Epuyén, where the 2018-2019 outbreak affected 34 individuals.
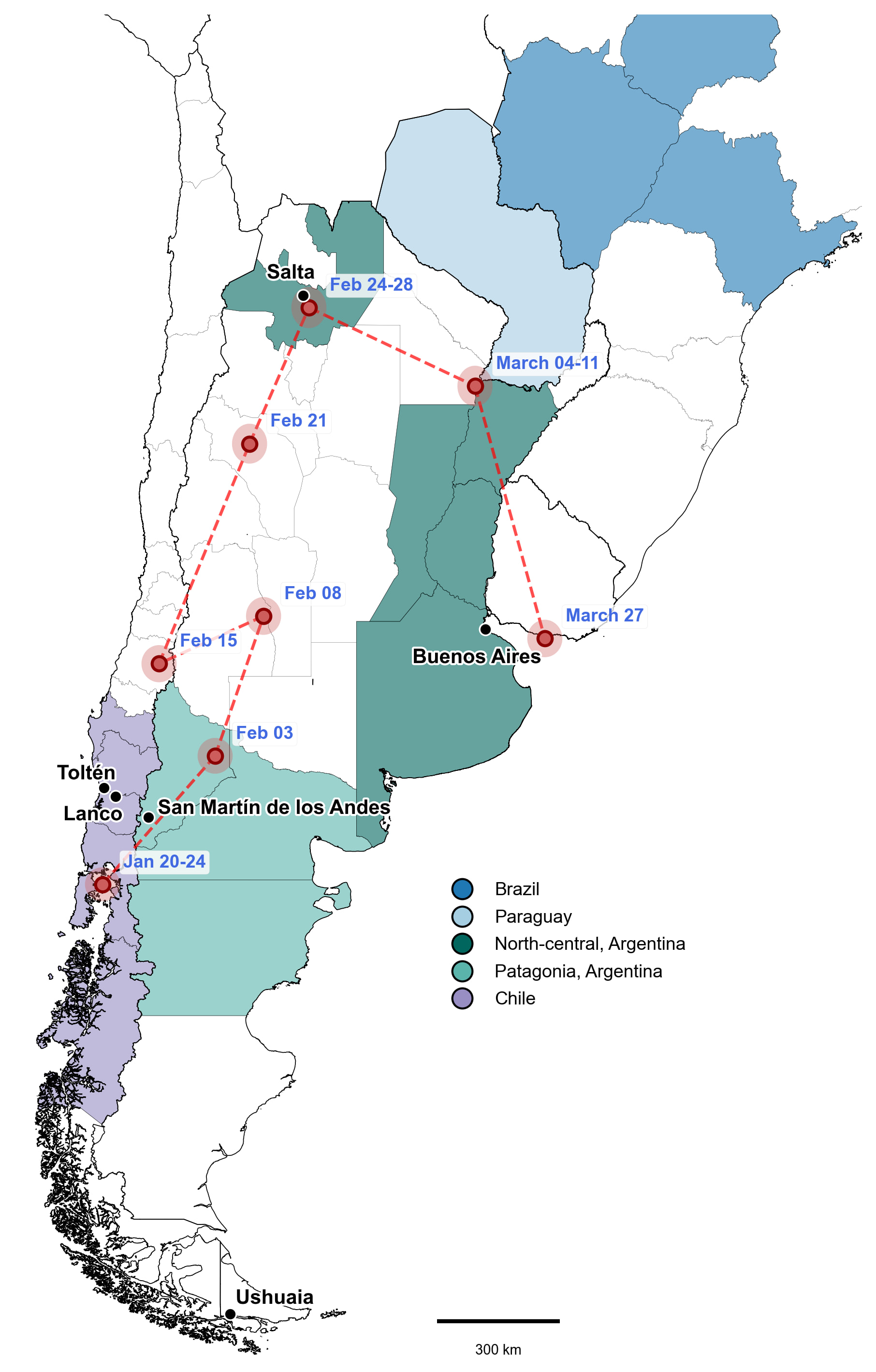
| Figure 4. Reported travel itinerary of the index case across southern south america in relation to the geographic distribution of O. andesense lineages. Macro-regions are colored associated with the phylogenetic clustering shown in Fig. 2. Red dashed lines indicate the reported travel itinerary and dates correspond to the approximate timing of travel between locations. Travel history was reconstructed from publicly available journalistic sources (Libération CheckNews, reporting by Brice Le Borgne, Eliette Pellissier, Alice Clair and Brice Borgne) |
Overall, while the available genomic data most strongly support a relationship with viruses circulating in the southern Chile-Argentina regions, the precise location and timing of exposure remain uncertain and should be interpreted cautiously pending additional epidemiological information and ongoing research. The relationship between Chilean rodent-derived sequences and the MV Hondius-associated genome, which is epidemiologically linked to Argentina, reflects the limited availability of rodent surveillance in the region. This highlights the need for improved genomic surveillance in the natural reservoir. Enhanced rodent-surveillance is key to understanding unobserved viral diversity in the natural reservoir, the drivers of viral circulation across borders and identifying lineages with outbreak potential. Concurrently, these findings raise fundamental ecological questions regarding the role of rodent host movement in the region, habitat connectivity, and ecological drivers that shape viral dynamics.
Disclaimer: Please feel free to download, share, use, and analyze these data. A preprint, unrelated to this blog post, with the Chilean rodent-derived sequences has been under review for publication since prior to the MV Hondius outbreak. These data are shared through Pathoplexus under its restricted-use policy, and we kindly ask users to adhere to those terms. If you intend to use these sequences or associated analyses for publication prior to the release of our paper, please contact us directly.
PP_00701B4.1, PP_00701C2.1, PP_00701FW.1, PP_00701GU.1, PP_00701HR.1, PP_00701JP.1, PP_00701KM.1, PP_00701D0.1, PP_00701EY.1
Authors:
- Ulloa-Zepeda, Lissette - Programa Hantavirus y Zoonosis, Universidad del Desarrollo Clínica Alemana
- Parker, Edyth - Department of Translational Medicine, The Scripps Research Institute, La Jolla, CA, USA; Institute of Genomics and Global Health, Redeemer’s University, Ede, Nigeria
- Andersen, Kristian - Department of Translational Medicine, The Scripps Research Institute, La Jolla, CA, USA
- Rambaut, Andrew - Institute of Ecology and Evolution, University of Edinburgh, Edinburgh, UK
- Vial, Cecilia - Programa Hantavirus y Zoonosis, Universidad del Desarrollo Clínica Alemana
Full phylogenetic trees: S segment, M segment, L segment
References
[1] Andes Hantavirus Outbreak on a Cruise Ship, 2026. N. Engl. J. Med. doi: 10.1056/NEJMc2606496 (2026)
[2]Martínez, V. P. et al. “Super-Spreaders” and Person-to-Person Transmission of Andes Virus in Argentina. N. Engl. J. Med. 383, 2230–2241 (2020).
[3]Alonso, D. O. et al. Person-to-Person Transmission of Andes Virus in Hantavirus Pulmonary Syndrome, Argentina, 2014. (2020).
[4]Padula, P. J. et al. Hantavirus Pulmonary Syndrome Outbreak in Argentina: Molecular Evidence for Person-to-Person Transmission of Andes Virus. Virology 241, 323–330 (1998).
[5]Experimental embedded trees for ANDV - Hantavirus. Virological Experimental embedded trees for ANDV (2026).
[6]Complete sequence of Orthohantavirus andesense virus: Swiss resident 2026 - Hantavirus. Virological Complete sequence of Orthohantavirus andesense virus: Swiss resident 2026 - #9 by emmahodcroft (2026).
[7]Palma, R. E., Boric-Bargetto, D., Torres-Pérez, F., Hernández, C. E. & Yates, T. L. Glaciation Effects on the Phylogeographic Structure of Oligoryzomys longicaudatus (Rodentia: Sigmodontinae) in the Southern Andes. PLOS ONE 7, e32206 (2012).
[8]Andreo, V., Glass, G., Shields, T., Provensal, C. & Polop, J. Modeling Potential Distribution of Oligoryzomys longicaudatus, the Andes Virus (Genus: Hantavirus) Reservoir, in Argentina. EcoHealth 8, 332–348 (2011).
[9]Vial, P. et al. Incubation Period of Hantavirus Cardiopulmonary Syndrome - Volume 12, Number 8—August 2006 - Emerging Infectious Diseases journal - CDC doi:10.3201/eid1208.051127.
[10]Brown S, Bhatia A, Shaju AM, Gabriel L, Hill J, Walker J and Carville S. Transmission parameters of hantavirus disease in human-to-human transmission contexts: a systematic evidence summary. UKHSA 2026