Stephen A Goldstein and Nels C Elde
University of Utah Department of Human Genetics
Salt Lake City, UT
Summary
SARS-CoV-2-like viruses remain sparsely sampled but are increasingly being discovered through analysis of both archived and newly collected samples from pangolins and bats. Newly discovered viruses are often screened for recombination events by comparing them to a subset of reference sequences, but only limited attempts have been made to use phylogenetics to capture an overall evolutionary picture of these viruses, and efforts to do rapidly become outdated. This post will be updated to reflect new insights provided as additional SARS-CoV-2 related sampling is reported. Most analysis focuses on the RdRp as a conserved gene in a region thought to be less recombination prone. In the case of SARS-CoV-2 itself, the most robust analyses do not support a role for recent recombination in its emergence [1, 2]. However, nucleotide identity analysis and gene-specific phylogeny reveals rampant recombination among SARSr-CoVs. Recombination may not only result in phylogenetic incongruence but also obfuscate evolutionary relationships when the recombination events are not easily identified. In the case of SARS-CoV-2-like viruses RmYN02 and RacCS203, an RdRp tree does not suggest a particularly close relationship. However, they share a spike recombination event, suggesting a very recent common ancestor. More broadly, recombination between the SARS-CoV-2 lineage and distant SARSr-CoV lineages, including the SARS-CoV lineage, is evident in the very recent past and presumably ongoing today. Our evolutionary understanding of these viruses will further improve with increased sampling and continued analysis of novel sequences in the lineage.
Findings
Continued sampling of archived and newly acquired bat samples have begun to populate the SARS-CoV-2 branch of SARSr-CoV, initially represented only by SARS-CoV-2 and RaTG13 [3]. These additional viruses include sequences from pangolins [4-6] and bats from southern China [7, 8] as well as viruses in Japan [9], Cambodia [10], and Thailand [11] thus extending the range of SARS-CoV-2-like viruses throughout east Asia. However, each discovery in isolation often fails to capture the entire picture of diversification in the evolutionary history of these viruses. This is unavoidable as publications are “frozen in time” whereas a constantly updated categorization is required with intensifying sampling specifically targeting the SARS-CoV-2-like viruses. The most recent such effort by Lytras et. al. [12] demonstrated that bat host ranges and recombination patterns suggest viral ranges are more expansive than previously appreciated. However, the study is limited by sparse sampling of related viruses.
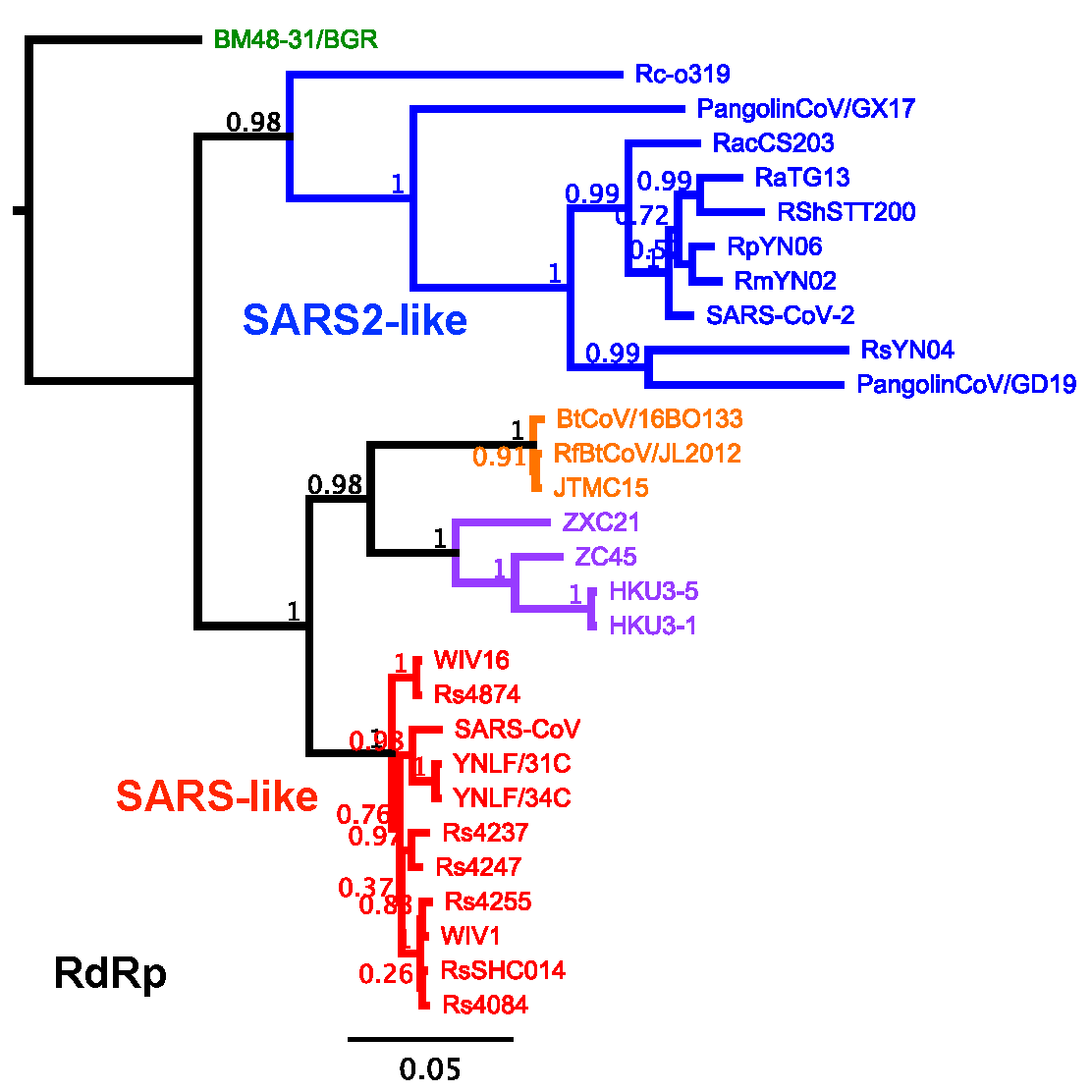
Despite renewed sampling efforts, these viruses remain lightly sampled relative to the SARS-CoV-like viruses as evidenced by extended branch lengths in maximum-likelihood phylogenetic analysis (Figure 1) and fewer representatives in absolute numbers. This RdRp phylogenetic tree represents SARSr-CoV diversity while discarding most nearly identical viruses in the SARS-CoV branch. Our RdRp analysis groups SARSr-CoVs into 4 or 5 lineages; a recent study classified five lineages [13] but it is unclear if the Zhoushan (ZXC21/ZC45)/HKU3 lineage (purple) and JL2012 lineage (orange) are sufficiently distinct to be categorized separately. Notably, in RdRp the SARS-CoV (red) and SARS-CoV-2 (blue) lineages are highly divergent with the SARS-CoV-2 lineage containing longer branch lengths indicating some combination of a more ancient origin and more sparse sampling. The closest relatives to SARS-CoV-2 in RdRp are RpYN06 and RmYN02 at >98% nucleotide identity whereas the southeast Asian viruses RacCS203 and RShSTT200 appear more distant and Rc-0319 sampled from Japan the most divergent member of the lineage, consistent with its geographic distance.
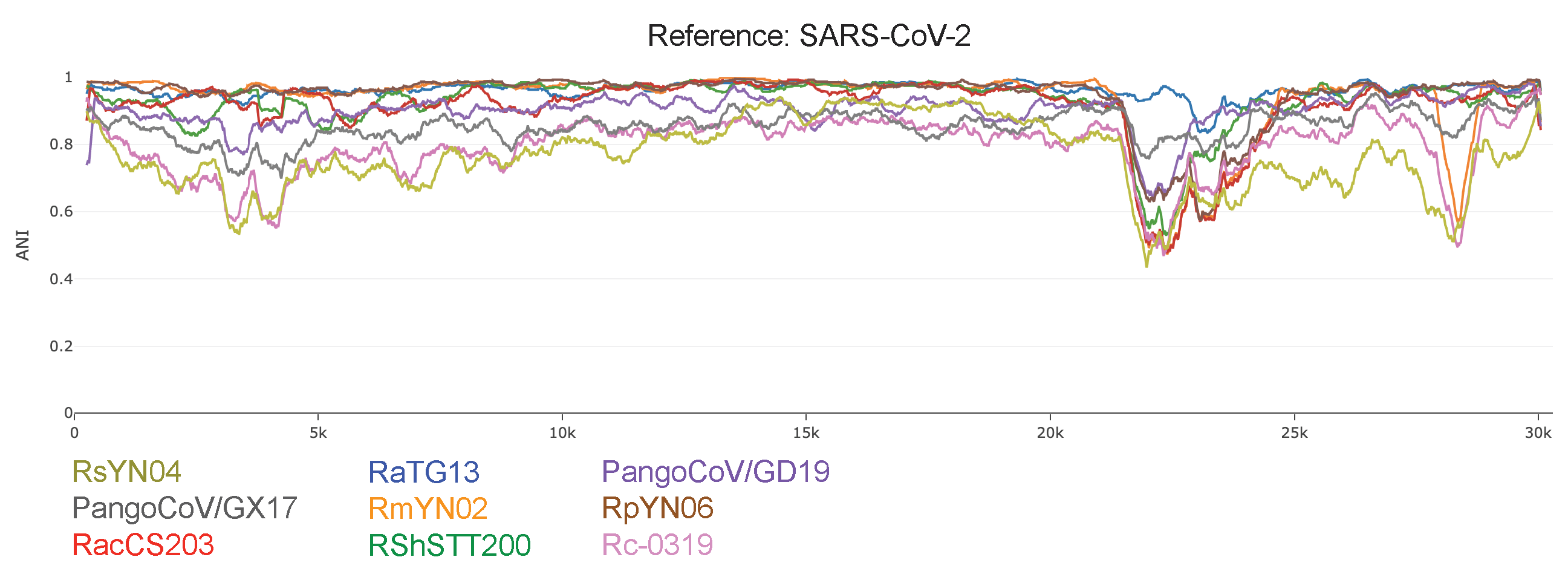
To better infer the influence of recombination in evolution of the SARS-CoV-2 lineage we conducted IDPlot [14] nucleotide identity analysis (Figure 2). Changes in average nucleotide identity (ANI) throughout the genome indicate that there is likely recombination in Orf1ab that is difficult to parse given the relative lack of diversity in this region. However, dramatic changes in ANI encompassing the spike and Orf8 genes provide more clearly defined signatures of recombination. Notably, the spike recombinant regions of RmYN02 and RacCS203 appear to perfectly overlap despite their distance on the RdRp tree. Newly discovered RsYN04 appears to have acquired substantial genetic information from a highly divergent lineage.
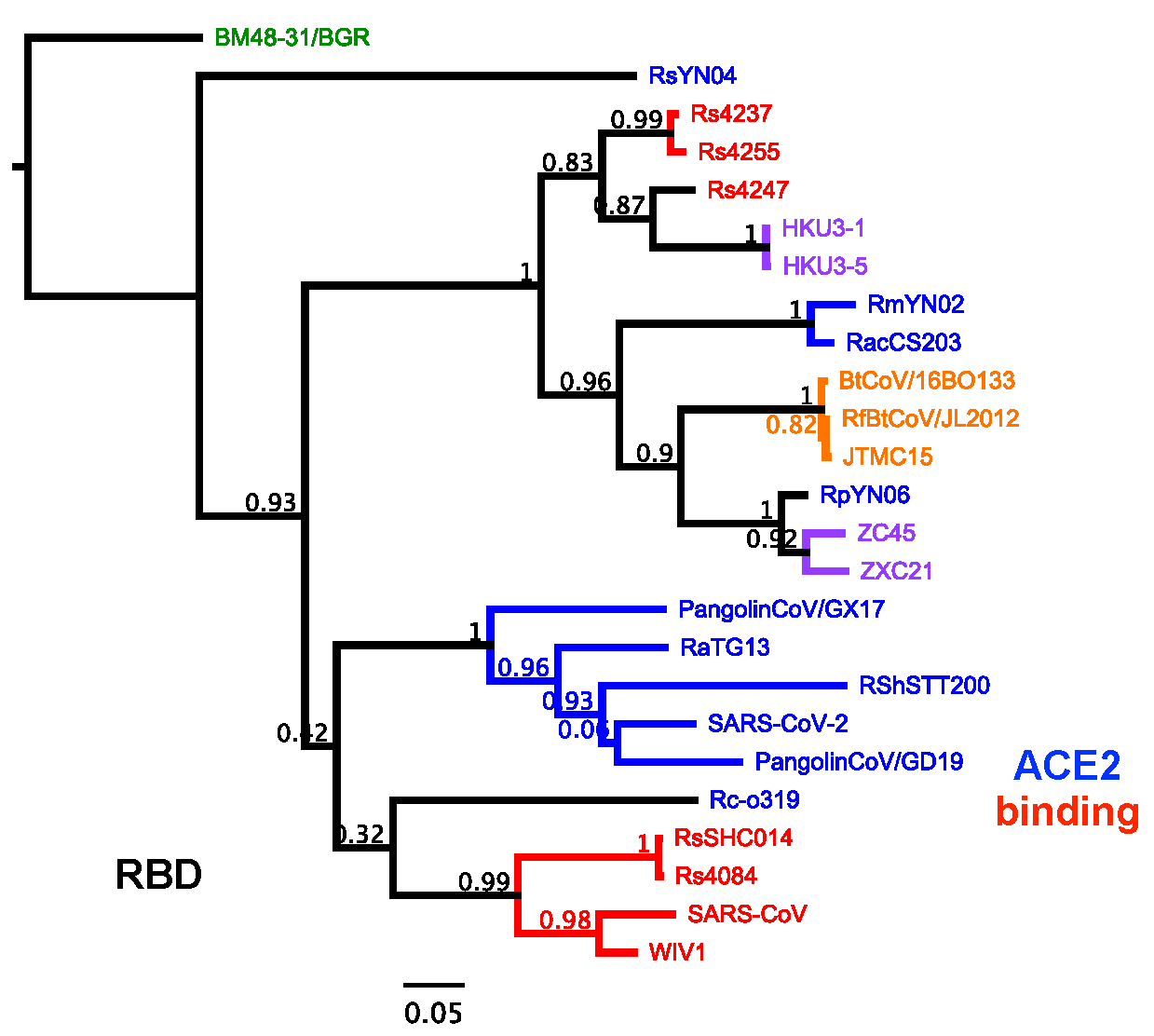
To better understand evolutionary relationships among spike sequences. we constructed a tree of the receptor binding domain (RBD). The most notable finding is that RmYN02 and RacCS203 group closely together, in contrast to the RdRp tree. Further analysis shows they are 95.5% identical in this region, the highest identity of any two SARS-CoV-2-like viruses in the dataset. One possibility is these viruses independently acquired RBDs from the same source. However, given the perfect overlap of their recombinant regions, a single recombination event subsequent to their divergence from the SARS-CoV-2 common ancestor but preceding their divergence from each other is a more parsimonious explanation. The divergence of RmYN02 from SARS-CoV-2 has been estimated at ~37 years ago [2], so this recombination event involving the RacCS203/RmYN02 spike almost certainly occurred even more recently. The acquisition by RmYN02 of a SARS-CoV-like Orf8 appears even more recent, and RacCS203 retains the ancestral SARS-CoV-2 Orf8 (Figure 4). The lack of a particularly close relationship in the RdRp suggests that after their split from a common ancestor, RacCS203 recombined back with SARS-CoV-2 and RaTG13-like viruses, akin to “back-crossing” of model organisms. These recombination events in Orf1ab appear frequent and difficult to detect as they likely involve small genomic regions among closely related viruses.
Notable in the RBD tree is the clustering of ACE-2-binding SARS-CoV-2 and SARS-CoV-like spikes, consistent with the recent description of their common ancestry and emergence of this RBD in the SARS-CoV lineage via recombination [13]. RsYN04, a newly discovered virus from Yunnan does not associate with any known viruses and is highly diverged, although its position is somewhat obscured given the convention of rooting the tree wth the European SARSr-CoV BM48-31/BGR sequence, from Bulgaria. The final major recombination event inferred by this tree involved RpYN06 which, based on RdRp phylogeny is the closest relative to SARS-CoV-2 and was recently identified in Yunnan province. However, its RBD groups closely with ZC45/ZXC21 and exhibits 94% nucleotide identity. These viruses were isolated from eastern China distant from the Yunnan sampling site of RpYN06 [12], suggesting closely related viruses have a broader geographic range, encompassing southern China, than previously appreciated.
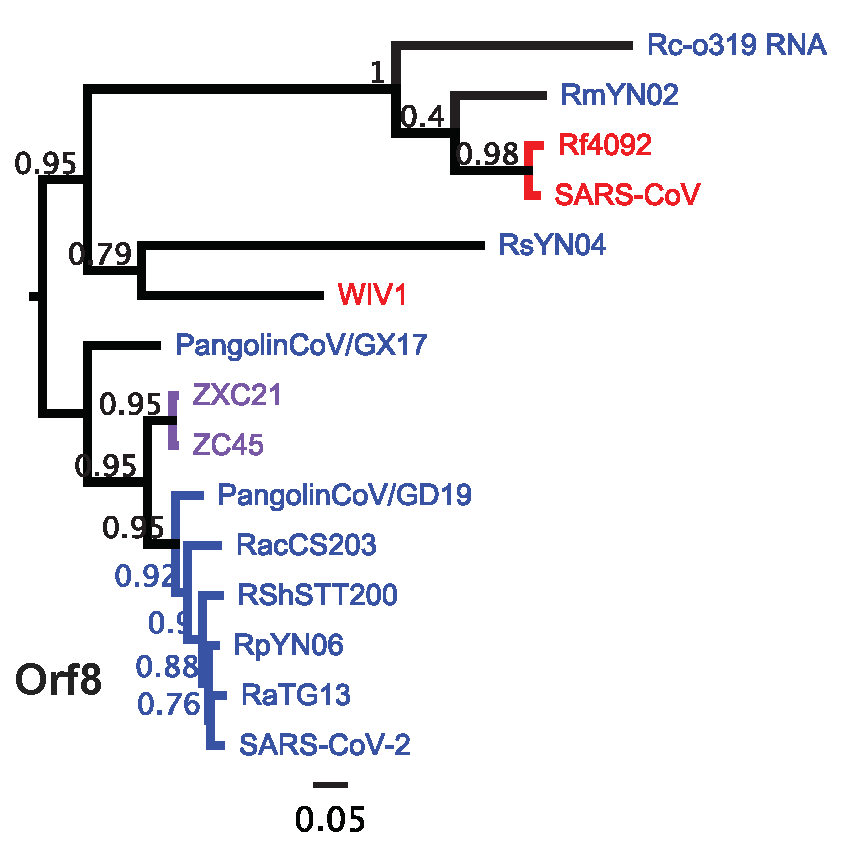
The final recombination hotspot in these viruses is in the 3’ Orf8 gene which appears highly dynamic in SARSr-CoVs [14-16]. Recombination here produced substantial diversity among the SARS-CoV-2 lineage Orf8 genes. Notably, RmYN02 and Rc-0319 group with the Orf8 sequences in the SARS-CoV lineage, albeit with considerable distance, while RsYN04 has less than 60% identity to any deposited Orf8 sequence although it may associate with WIV1 in this dataset.
Conclusion
SARS-CoV-2-like viruses remain sparsely sampled and therefore a detailed. understanding of the evolutionary history of these viruses remains elusive. However, improved sampling over the last several months is advancing and is essential to better understand the origins of SARS-CoV-2 and the evolution of related viruses. Accounting for recombination is vital for providing an accurate picture of the relatedness between these viruses. Increasingly sophisticated recombination detection programs can identify candidate regions defined by breakpoints but have limited ability to detect “minor” recombination events involving closely related viruses, which hinders making accurate phylogenetic inferences at branch tips. Additional information such as shared recombination events can supplement recombination analyses and strengthen inferences of evolutionary history. This post will be updated as warranted by the discovery of novel SARS-CoV-2-like viruses.
- Boni, M.F., et al., Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nature Microbiology, 2020. 5(11): p. 1408-1417.
- Wang, H., L. Pipes, and R. Nielsen, Synonymous mutations and the molecular evolution of SARS-CoV-2 origins. Virus Evolution, 2021. 7(1).
- Zhou, P., et al., A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 2020. 579(7798): p. 270-273.
- Liu, P., W. Chen, and J.-P. Chen, Viral Metagenomics Revealed Sendai Virus and Coronavirus Infection of Malayan Pangolins (Manis javanica). Viruses, 2019. 11(11): p. 979.
- Lam, T.T.-Y., et al., Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature, 2020. 583(7815): p. 282-285.
- Xiao, K., et al., Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature, 2020. 583(7815): p. 286-289.
- Zhou, H., et al., A Novel Bat Coronavirus Closely Related to SARS-CoV-2 Contains Natural Insertions at the S1/S2 Cleavage Site of the Spike Protein. Current Biology, 2020. 30(11): p. 2196-2203.e3.
- Zhou, H., et al., Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. 2021, Cold Spring Harbor Laboratory.
- Murakami, S., et al., Detection and Characterization of Bat Sarbecovirus Phylogenetically Related to SARS-CoV-2, Japan. Emerging Infectious Diseases, 2020. 26(12): p. 3025-3029.
- Hul, V., et al., A novel SARS-CoV-2 related coronavirus in bats from Cambodia. 2021, Cold Spring Harbor Laboratory.
- Wacharapluesadee, S., et al., Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nature Communications, 2021. 12(1).
- Lytras, S., et al., Exploring the natural origins of SARS-CoV-2. 2021, Cold Spring Harbor Laboratory.
- Wells, H.L., et al., The evolutionary history of ACE2 usage within the coronavirus subgenus Sarbecovirus. Virus Evolution, 2021. 7(1).
- Goldstein, S.A., et al., Extensive recombination-driven coronavirus diversification expands the pool of potential pandemic pathogens. 2021, Cold Spring Harbor Laboratory.
- Lau, S.K.P., et al., Severe Acute Respiratory Syndrome (SARS) Coronavirus ORF8 Protein Is Acquired from SARS-Related Coronavirus from Greater Horseshoe Bats through Recombination. Journal of Virology, 2015. 89(20): p. 10532-10547.
- Hu, B., et al., Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLOS Pathogens, 2017. 13(11): p. e1006698.
Acknowledgements
Particular thanks are in order to Edward C. Holmes who provided sequences associated with his and collaborators’ recent preprint describing newly identified bat coronaviruses. Zhou et. al.(8)