On September 4, 2025, the DRC Ministry of Health officially declared the 16th EVD outbreak in DRC. This new outbreak is described as a new spillover (see earlier post) and to date (September 7, 2025), 14 deaths have been noted among 45 suspected cases. Among the 5 EBOV PCR positive samples, 4 genomes have been generated.
Four genomes are available on the Pathoplexus.org EBOV database. See this post for details about the sample handling and sequencing. The consensus genomes were generating using the ARTIC amplicon pipeline – amplicon-nf.
Table 1 | Sampled cases
| ID | Pathoplexus accession | Sample type | Collection date | Status on collection | Age range (years) | sex |
|---|---|---|---|---|---|---|
| 25FHV168 | PP_003RXEP | Whole blood | 2025-09-01 | Alive | <5 | F |
| 25FHV170 | PP_003RXFM | Whole blood | Sep-2025 | Alive | unknown | M |
| 25FHV172 | PP_003RXGK | Whole blood | 2025-08-29 | Alive | <5 | M |
| 25FHV173 | PP_003RXHG | Oral swab | 2025-09-01 | diseased | >25 | F |
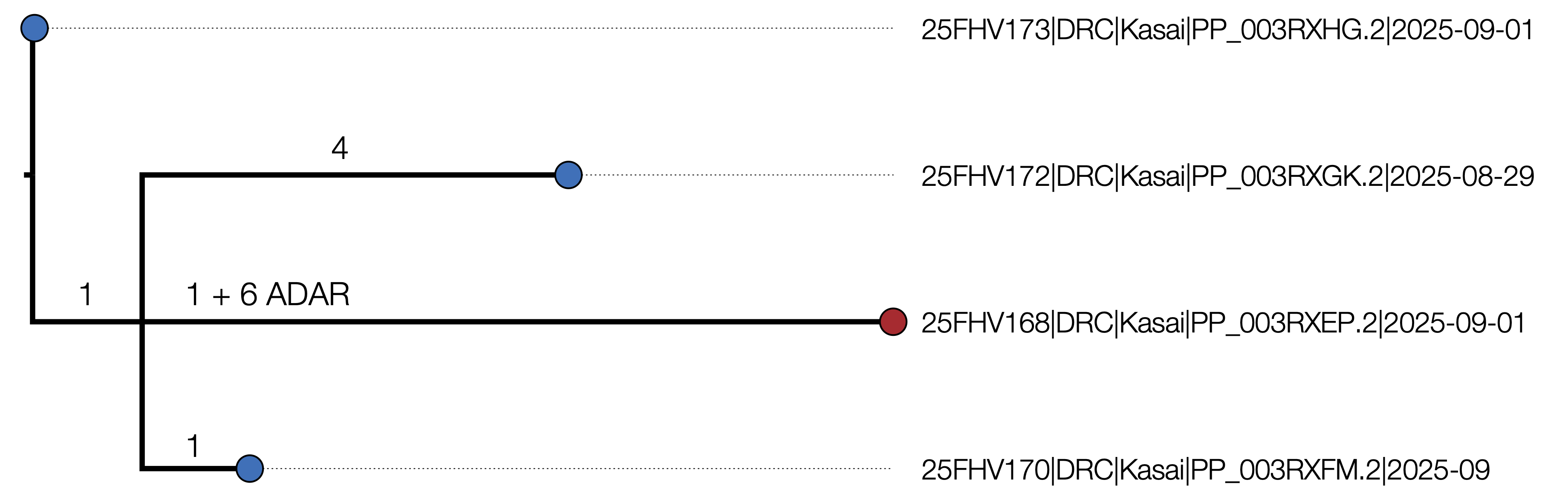
Genome 25FHV168 (Pathoplexus accession PP_003RXEP) exhibits six T->C changes relative to the other genomes and the EBOV reference genome in a tight cluster – within a 101 nucleotide stretch starting at site 2867. Such a pattern has previously been identified and ascribed to the action of ADAR. These mutations are in an intergenic region between the NP and VP35 genes so probably do not affect the virus fitness (or we would not observe them).
The remaining 16 mutations within the outbreak genomes are listed in Table 2. Two of the inferred mutations are phylogenetically incompatible with each other and may represent a homoplasy or reversion or may be a sequencing error.
Table 2 | Mutations in the outbreak genomes relative to reference
| Genomes | Site | Mutation | Notes | |
|---|---|---|---|---|
| 25FHV170 | 340 | A->G | 5’ UTR | |
| 25FHV172 | 8328 | A->G | intergenic region | |
| 25FHV168, 170, 172 | 9258 | G->A | VP30 synonymous | * |
| 25FHV168 | 10009 | T->C | intergenic region | |
| 25FHV172 | 11536 | C->T | intergenic region | |
| 25FHV168, 170, 173 | 12351 | A->G | L synonymous | * |
| 25FHV172 | 18835 | A->G | 3’ UTR |
* these mutations are phylogenetically conflicting and may be the result of a convergent mutation, a reversion or a sequencing error.
An initial maximum likelihood phylogenetic tree shows some structure (Figure 1) with a maximum of 5 nucleotide differences between the most divergent pairs.
A)
B)
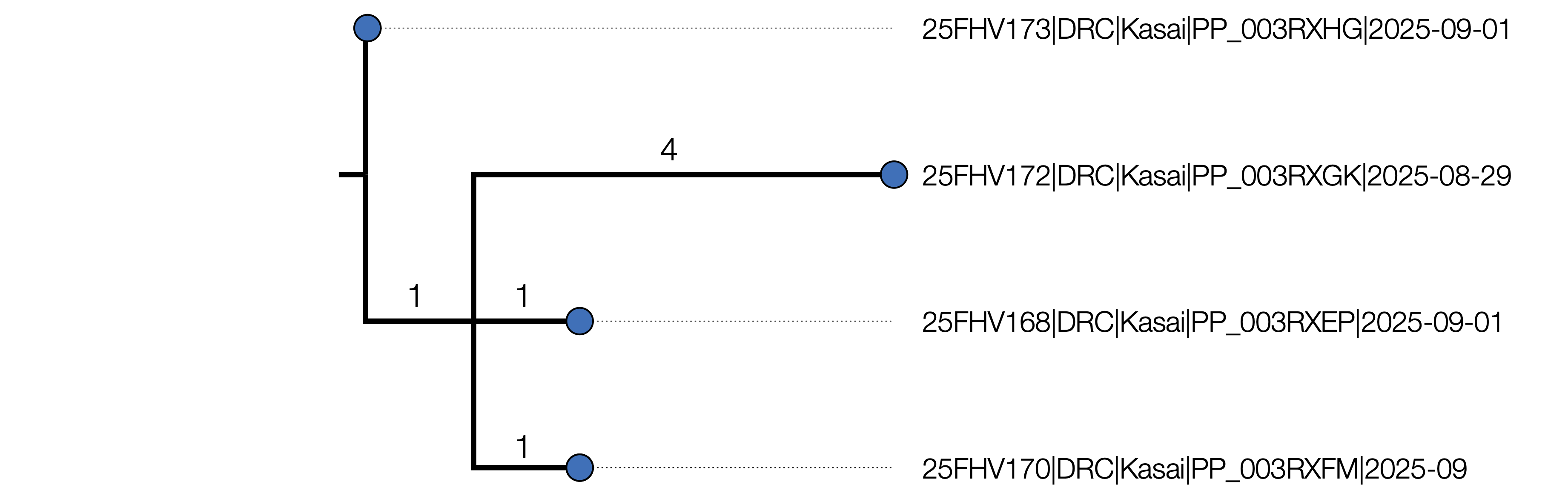
Figure 1 | Maximum likelihood tree of the 4 outbreak genomes inferred using IQTREE2 [1]. It is rooted using the EBOV reference genome (accession NC_002549, DRC/Yambuku-Mayinga/1976) which is also the genetically closest genome to the outbreak. The branch labels represent the inferred number of nucleotide changes on each branch and the node labels are Ultrafast Bootstrap support values. A) 25FHV168 (red) including 6 putative ADAR mutations. B) The tree with the ADAR mutations masked out.
Temporal analysis
Although there are only four genomes available at present, we have performed a time-scaled phylogenetic analysis using BEAST [2] in order to estimate whether the start of the outbreak significantly pre-dates the first detection.
For the purposes of these analyses, the six putative ADAR mutations in genome 25FHV168 have been masked out as they will distort the phylogenetic and temporal signal.
To perform this analysis we required a fixed rate of evolution due to the limited temporal range of sampling. A comprehensive review [3] compiled estimates from the literature for the 2014-2016 epidemic with values between ~1.9 × 10-3 [95% Bayesian credible interval: 1.11, 2.91 × 10-3] subst/site/year for an early period of the epidemic [4] to ~1.2 × 10-3 [1.13, 1.27 × 10-3] across all public data. To explore this range we use a rate of 1.0 x 10-3 and 2.0 x 10-3 as the upper and lower bounds of these estimates. We also used a constant size coalescent model and an exponential growth coalescent model for both rates (Table 3).
Table 3 | Time to most recent common ancestor (tMRCA) estimates
| Coalescent model | Assumed Rate substs/site/year |
Mean tMRCA | Lower 95% HPD | Upper 95% HPD |
|---|---|---|---|---|
| Constant | 1.0 x 10-3 | 2025-07-14 | 2025-06-05 | 2025-08-17 |
| Constant | 2.0 x 10-3 | 2025-08-08 | 2025-07-19 | 2025-08-25 |
| Exponential | 1.0 x 10-3 | 2025-07-24 | 2025-06-25 | 2025-08-19 |
| Exponential | 2.0 x 10-3 | 2025-08-11 | 2025-07-26 | 2025-08-24 |
Observations
With few genomes and the need to make a strong assumption about the rate of evolution, these estimates should be considered illustrative at this stage. Although the mean estimates for the slower rate are older than the current understanding about the timeline of the outbreak, the 95% HPDs for all the runs include late August.
Further epidemiological work is being undertaken that may be informative about the early cases.
Statement on continuing work and analyses prior to publication
Please note that this data is based on work in progress and should be considered preliminary. Our analyses are ongoing and a publication communicating our findings is in preparation.
Four genomes are now available on Pathoplexus.org. These are publicly accessible under the Pathoplexus ‘Restricted’ licence and we would be grateful if the terms of this were respected.
If you intend to use our data prior to our publication, please contact Prof. Placide Mbala-Kingebeni.
Authors
Key contributors to data collection/molecular testing/data interpretation/whole genome sequencing/bioinformatics analysis/phylogenetic analysis, and manuscript writing):
Adrienne Amuri-Aziza (INRB, Kinshasa, DRC)
Gradi Luakanda-Ndelemo (INRB, Kinshasa, DRC)
Jean-Claude Makangara-Cigolo (INRB, University of Kinshasa, Kinshasa, DRC; Institute of Social and Preventive Medicine, University of Bern, Bern, Switzerland)
Prince Akil-Bandali (INRB, Kinshasa, DRC)
Servet Kimbonza (INRB, Kinshasa, DRC)
Fiston Cikaya-Kankolongo (INRB, Kinshasa, DRC)
Princesse Paku-Tshambu (INRB, Kinshasa, DRC)
Elzedek Mabika-Bope (INRB, Kinshasa, DRC)
Emmanuel Lokilo (INRB, Kinshasa, DRC)
André Citenga (INRB, Kinshasa, DRC)
Raphael Lumembe (INRB, University of Kinshasa, Kinshasa, DRC)
Gabriel Kabamba (INRB, University of Kinshasa, Kinshasa, DRC)
Christian Ngandu (Institut National de Santé Publique (INSP), Kinshasa, DRC)
Louis Tshulo (Division Provinciale de la Santé, Kasaï, DRC)
Mathias Mossoko (Institut National de Santé Publique (INSP), Kinshasa, DRC)
Dieudonné Mwamba (Institut National de Santé Publique (INSP), Kinshasa, DRC)
Elisabeth Pukuta (INRB, Kinshasa, DRC)
Eddy Kinganda-Lusamaki (INRB, University of Kinshasa, Kinshasa, DRC; TransVIHMI, Université de Montpellier, INSERM, IRD, Montpellier, France)
Andrew Rambaut (University of Edinburgh, UK)
Sam Wilkinson (University of Birmingham, UK)
Áine O’Toole (University of Edinburgh, UK)
Nicholas Loman (University of Birmingham, UK)
Josh Quick (University of Birmingham, UK)
Daniel Mukadi-Bamuleka (INRB, University of Kinshasa, Laboratoires P2/P3 Rodolphe -Merieux INRB-Goma, DRC)
Patrick Mukadi (INRB, University of Kinshasa)
Dieudonné Mumba Ngoyi (INRB, University of Kinshasa, Kinshasa, DRC)
Steve Ahuka-Mundeke (INRB, University of Kinshasa, Kinshasa, DRC)
Jean-Jacques Muyembe-Tamfum (INRB, University of Kinshasa, Kinshasa, DRC)
Tony Wawina-Bokalanga (INRB, University of Kinshasa, Kinshasa, DRC; Department of Clinical Sciences, Institute of Tropical Medicine, Antwerp, Belgium)
Placide Mbala-Kingebeni (INRB, University of Kinshasa, Kinshasa, DRC; South African National Bioinformatics Institute, University of the Western Cape, South Africa)
Acknowledgements
We are grateful to the ARTIC Network ( ARTIC network | ARTIC network - pathogen genomics from sample to response ) and the University of Nebraska for the provision of the primers. The biofire panels and primers were provided by Culmen International under their cooperative Agreement funded by the US CDC. We also acknowledge the Africa Pathogen Genomic Initiative, Agence Française de Dévelopement through the AFROSCREEN project (grant agreement CZZ3209, coordinated by ANRS-MIE Maladies infectieuses émergentes in partnership with Institut de Recherche pour le Développement (IRD) and Pasteur Institute) ; the Global Funds; the Belgian Directorate-General for Development Cooperation and Humanitarian Aid; the Research Foundation Flanders (“Fonds voor Wetenschappelijk Onderzoek–Vlaanderen”); Institute of Tropical Medicine Structurele Onderzoeksfinanciering (Flemish Government; Science, Technology, and Innovation); the Wellcome Trust (206298/Z/17/Z ARTIC and 313694/Z/24/Z ARTIC 2) and the World Health Organization for their support to genomic surveillance efforts in DRC through equipment acquisition, reagents procurement and logistic support.
Collaborating institutions and agencies
Africa Centres for Disease Control and Prevention, Addis Ababa, Ethiopia
Biosurv international
Culmen International
Institute of Ecology and Evolution, University of Edinburgh, Edinburgh, UK
Institute of Tropical Medicine, Antwerp, Belgium
TransVIHMI, Université de Montpellier, INSERM, IRD, Montpellier, France
University of Birmingham, Birmingham, UK
University of California Los-Angeles (UCLA), Los-Angeles, USA
University of Manitoba, Winnipeg, Manitoba, Canada
University of Bern, Switzerland
US Department of Agriculture, Manhattan, KS, USA
Viral Special Pathogens, US Centers for Disease Control and Prevention, Atlanta, GA, USA
World Health Organization Country Office, Kinshasa, Democratic Republic of the Congo
World Health Organization, Geneva, Switzerland
References
1. Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, et al. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol. 2020 May 1;37(5):1530-4.
2. Baele G, Ji X, Hassler GW, McCrone JT, Shao Y, Zhang Z, et al. BEAST X for Bayesian phylogenetic, phylogeographic and phylodynamic inference. Nat Methods. 2025;22: 1653–1656.
3. Holmes EC, Dudas G, Rambaut A, Andersen KG. The evolution of Ebola virus: Insights from the 2013-2016 epidemic. Nature. 2016;538: 193–200.
4. Gire SK, Goba A, Andersen KG, Sealfon RSG, Park DJ, Kanneh L, et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science. 2014;345: 1369–1372.