Artem Babaian
[1] University of Cambridge, Cambridge, UK
[2] St. Edmund’s College, Cambridge, UK
Application Link: palmID
Source: palmID github (GPLv3)
Introduction
The rapid growth of nucleic acid sequencing has driven an exponential expansion in novel viruses with a vast sequence diversity which precludes efficient classification by traditional (human-curated) taxonomic means (Edgar et al., 2022). High-throughput description of viruses can be achieved through the use of hallmark genes for taxa resolution, which for RNA viruses is the RNA-dependent RNA Polymerase (RdRP) gene (Wolf et al., 2018). We recently proposed RdRP ‘palmprint’ barcode as a structurally well-defined sub-sequence of the polymerase palm sub-domain delineated by conserved catalytic motifs A, B, and C. These sequence motifs which delineate the ‘palmprint’ are rapidly detected with the program palmscan, and a database of RdRP palmprints exists called PALMdb (Babaian and Edgar, 2021). Together autonomous detection and analysis of palmprints can be deployed to create a meta-data aggregation platform to achieve a data-driven description of virus ecology.
palmID (www.serratus.io/palmid) is a freely-available web tool for the procedural analysis of an input RdRP containing viral sequence (nucleotide or amino acid). This tool offers a proof-of-concept workflow for high-throughput interpretation of novel RNA viruses, paving the path for the continued ultra-rapid growth in RNA virus discovery anticipated in the coming decade (Edgar et al., 2022, Zayed et al., 2022, Neri et al., 2022).
Human Rubella virus (Rubivirus rubellae) is a highly contagious human pathogen which causes “German measles” and has been implicated in encephalitis and congenital birth defects following maternal infection (Cooper. 1985). Two non-human Rubiviruses were recently described: Ruhugu (Rubivirus ruteetense) infecting cyclops leaf-nosed bats sampled in Uganda, and Rustrela (Rubivirus strelense) infecting yellow-necked field mice in Germany. Rustrela virus was identified after causing acute encephalitis in a donkey, capybara, and Bennett’s tree kangaroo, demonstrating that rubiviruses present a risk of zoonotic spillover in mammals (Bennet et al., 2020). Surveillance of the genetic diversity, geographic- and host-ranges of Rubivirus is thus warranted in anticipation of future spill-over.
palmID: web-application for exploring the RNA virosphere
The known virome is growing and modern computational virology infrastructure should anticipate the integration of viral sequences and meta-data to number in the billions of records by the end of the decade. To demonstrate the functional application of palmprints for the aggregation of sequences and meta-data, I created palmID (Serratus), a free web-analysis tool (also available as a downloadable container) which receives a known or novel RdRP sequence as input and aggregates sequence and meta-data from similar viral RdRP (Figure 1).
Figure 1. Data-flow in palmID
Overview of the palmID workflow. A user-input RNA dependent RNA Polymerase (RdRP) sequence is analysed with palmscan and aligned against all known palmprints in PALMdb (v20210314, Babaian and Edgar, 2021). Each input-PALMdb palmprint match is weighted by its global-alignment amino-acid identity. Sequencing libraries within the Sequence Read Archive (SRA) (Edgar et al., 2022) are indexed against all species-like operational taxonomic units (sOTU) in PALMdb, thus the meta-data in the SRA can be aggregated for all input-PALMdb matches to generate a viral web-report for the input sequence.
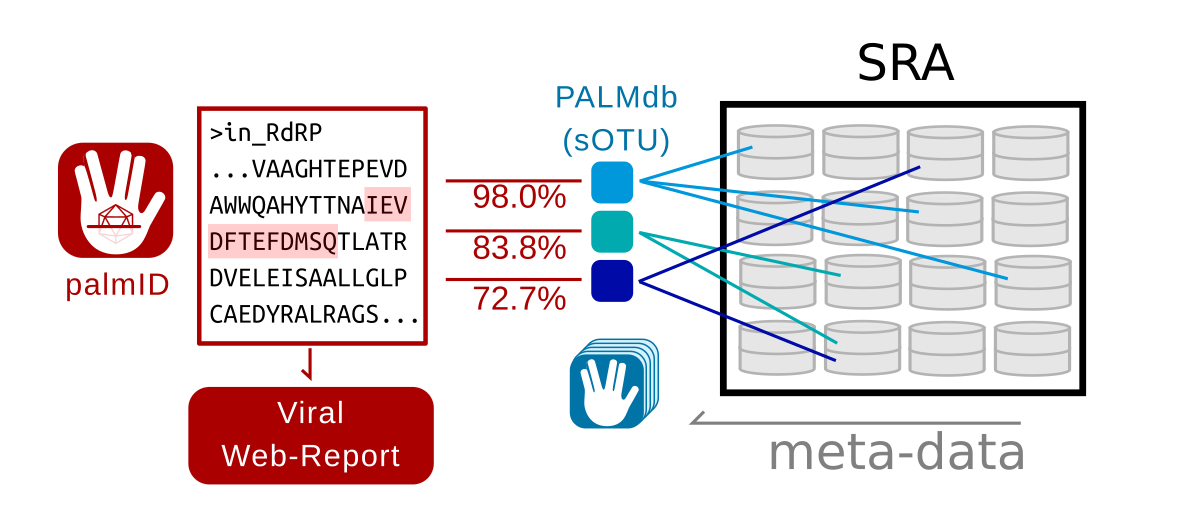
In brief, palmID implements palmscan (Babaian and Edgar, 2021) for the detection of a valid palmprint sub-sequence (Figure 2), and as a quality-control measure the input palmprint score, and length distribution are compared against a reference set of 15,000 canonical RdRP sequences from GenBank (Figure 3). The user-input palmprint is then aligned against all PALMdb sOTU centroids with DIAMOND (–ultra-sensitive -e 0.00001, Buchfink et al., 2021) to retrieve the set of matching palmprints (upto 500 hits), with matching palmprints coarsely grouped into “species-like” (>90% identity), “genus-like” (70-90%), “family-like” (45-70%), or else “phylum-like” matches (Extended Data Fig. 4d). Using the Serratus API, each palmprint match is queried against RdRPs identified from the Sequence Read Archive (SRA) to retrieve the set of sequencing runs containing input-palmprint or neighbours. Corresponding meta-data are then aggregated. For example, geospatial distribution, sample annotation and virus-associated organisms via K-mer classification (retrieved from NCBI STAT, Katz et al., 2021) are displayed (Figure 3).
Interactive figures of this data are generated, and raw data-tables are available for download to the user with a typical running time of around two minutes.
Figure 2. Rubella Virus palmprint motifs
Example palmscan report using the Rubella virus non-structural polyprotein as input (accession: NP_062883.2). The 99-aa palmprint sub-sequence is extracted and assigned a quality score, along with each motif.
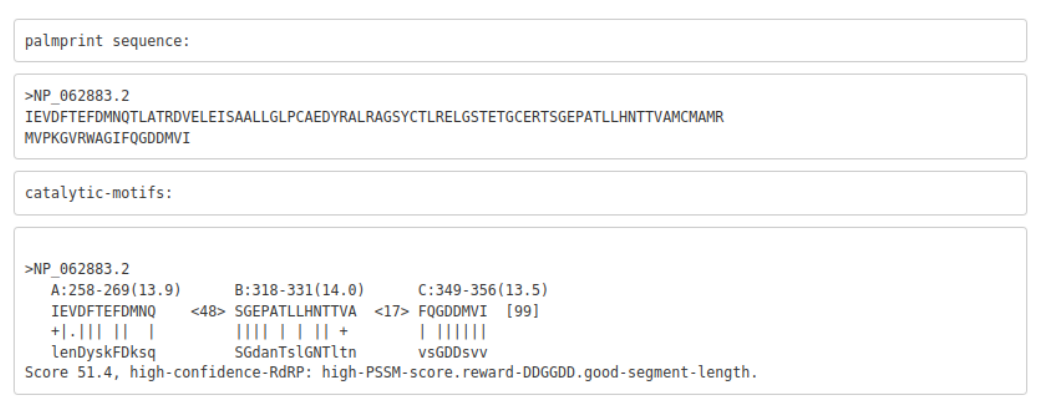
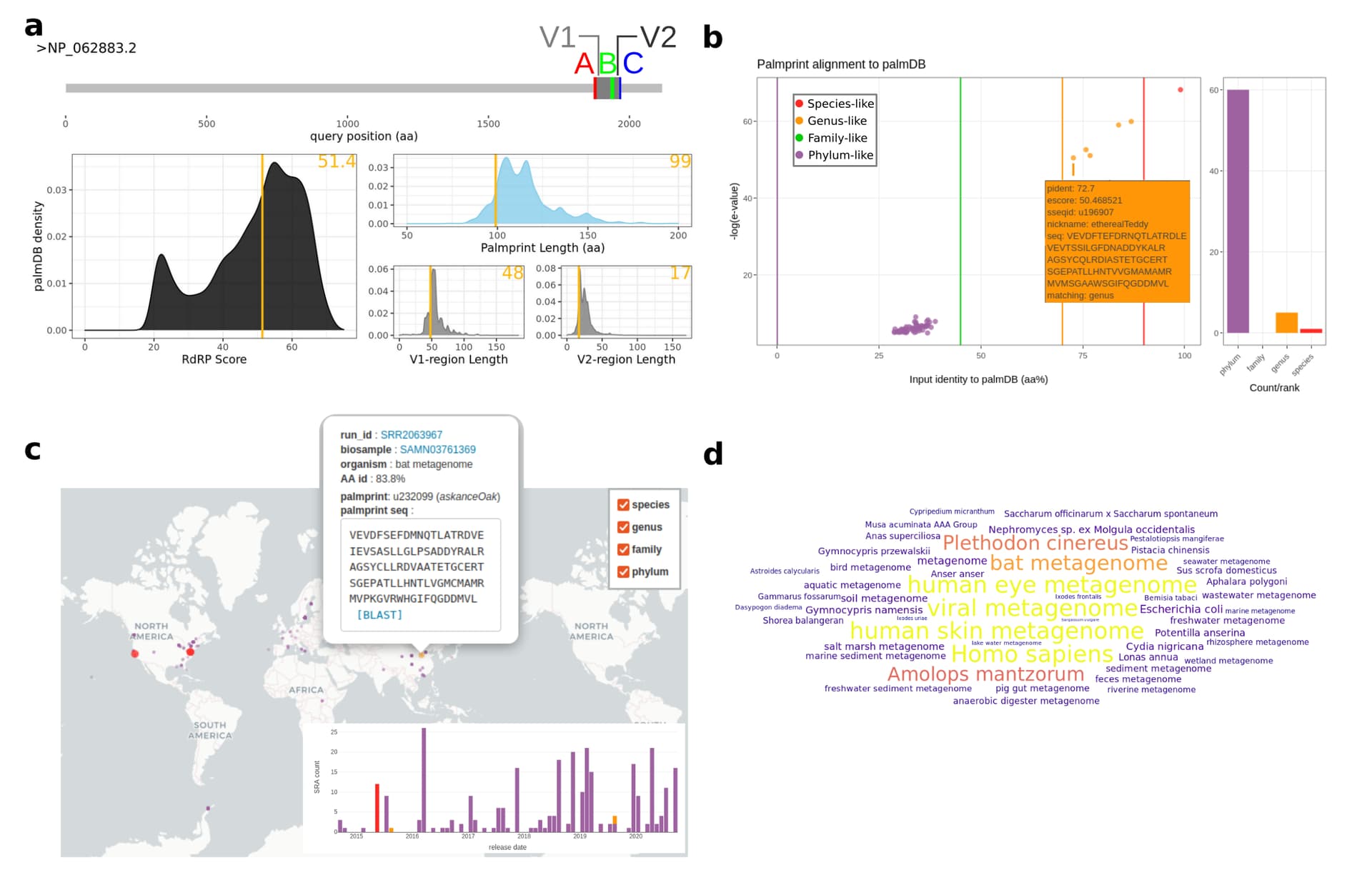
Figure 3. Rubella Virus palmID Viral Report
Select procedurally generated figures from the palmID web-report for Rubella virus (Full Online Report). A Quality control assessment of the input-palmprint displays palmprint-subsequence coordinates within input-sequence, and input score and component lengths compared against a reference set of 15,000 GenBank RdRP sequences (PALMdb v20210302). B Interactive input-palmprint identity and e-value (negative log10) graph reporting 66 matching palmprints from PALMdb (average aa-identity of 37.9%, range 28.7-99%) and MUSCLE multiple sequence alignment (not shown). Hits are coarsely striated into species-like to phylum-like categories. C Matching palmprints are identified in 261 distinct sequencing runs in the SRA from which meta-data are aggregated. Release-date timeline of all samples (inlay) and geospatial coordinates in the 201/261 (77%) available runs are plot on an interactive world-map with a one-click BLAST link available. D Word-cloud of the ‘organism’ data field from the sequencing runs, with size-colour scaled by the input-PALMdb palmprint alignment weighting (percent identity) and organism-level K-mer classification taken from STAT (Katz et al., 2021, not shown). High-identity Rubella virus palmprints are seen in libraries annotated asviral metagnome,human skin metagenome,Homo sapiens, while more distant palmprints are seen in libraries annotated asbat metagenome,Amolops mantzorumandPlethodon cinereus(Supplementary Table 1).
Ruche, Rumple, and Ruffle, Recent Rubivirus Relatives
To exemplify the utility of palmprints in navigating RNA viruses, I sought to identify new Rubiviruses using the palmID web interface. palmID identifies palmprints with Palmscan, searches PALMdb (Babaian and Edgar, 2021) and aggregates virus-associated meta-data from the Sequence Read Archive (Edger et al., 2022) into a user-facing report (Methods). The result of a two-minute search (results: Serratus) reported five genus-like virus sequence matches to Rubella: Ruhugu (86.9% palmprint identity to Rubella), Rustrela (76.8%) and three uncharacterised viruses (Figure 4).
Figure 4. Rubi- and rubi-like viruses identified by palmID
A Genome synteny among of Rubiviruses (RV) and related Matonaviruses (MV) showing significant (< E-04) protein domain matches detected by hmmscan. B Parallel phylogenetic tree (IQtree2) created from the common RNA dependent RNA polymerase (RdRP) sequences or concatenated capsid and E2/E1 glycoproteins, inlay showing unrooted RdRP-tree. C Protein sequence alignment of the common RdRP fragment with motif A,B, and C highlighted.
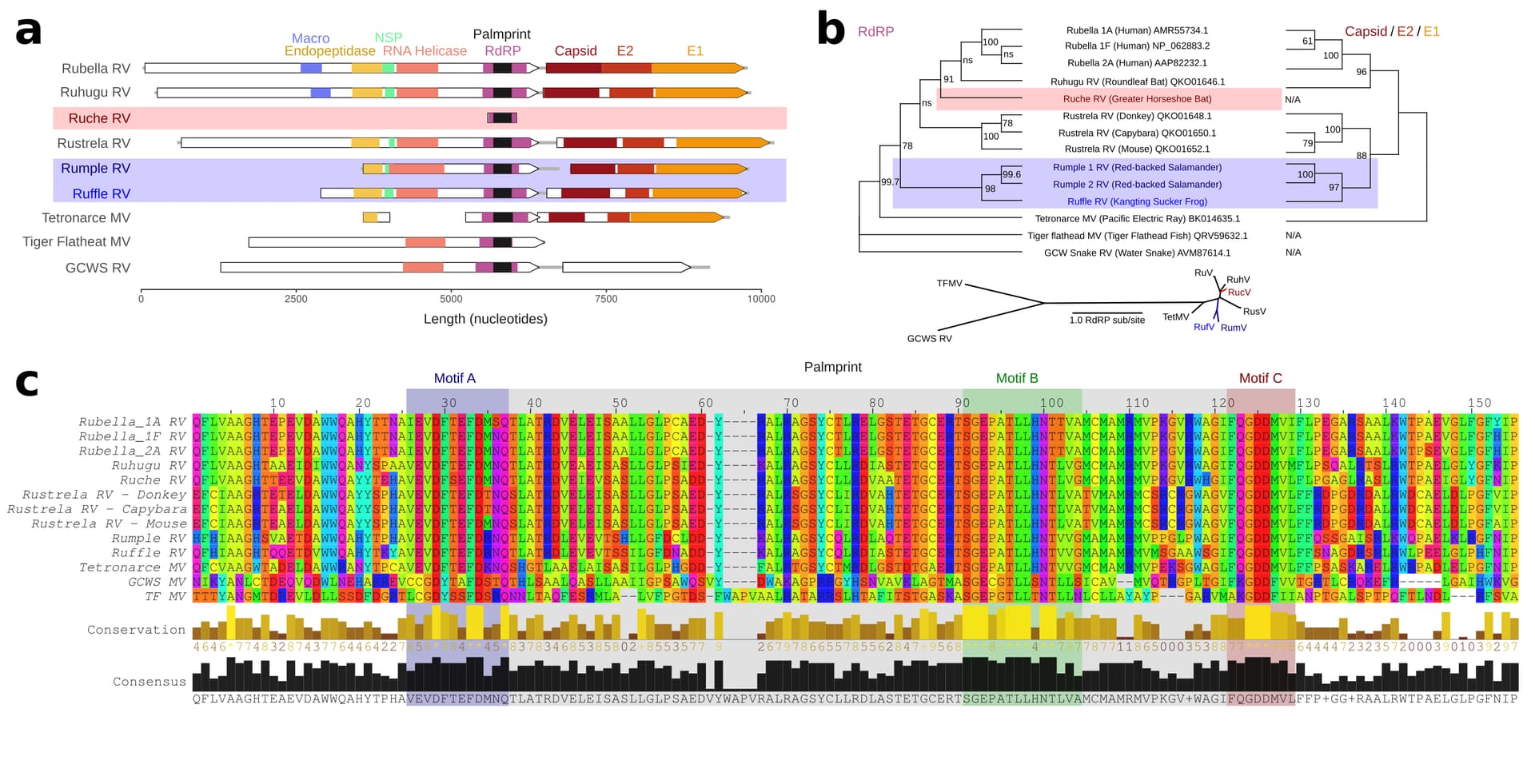
I refer to the novel viruses as Ruche Rubivirus (83.8%), Rumple Rubivirus (75.8%), and Ruffle Rubivirus (72.7%), respectively (Extended Table 1) and assembled the libraries reported to contain these palmprints (Fig 3). Ruche virus was identified from an RdRP fragment in a sample annotated as originating from a Greater Horseshoe Bat (Rhinolophus ferrumequinum) collected in 2013 in Shanxi, China (Wu et al., 2016). Rumple virus was identified in four samples originating from a North American Red-backed Salamander (Plethodon cinereus) experimentally housed and infected with chytrids (Ellison et al., 2020), and Ruffle virus was observed in Kangting Sucker frog (Amolops mantzorum) sampled no later than 2017 in Sichuan, China (Xia et al., 2018). While these assemblies expand the known diversity of Rubi-like viruses, the primary significance of the latter results is a demonstration that palmprint-based classification of RNA viruses enables rapid search and aggregation of virus-associated meta-data from large databases including geospatial locations, host-organism associations and virus phylogeny.
Conclusion
Public sequencing databases are growing exponentially, doubling ever ~18 months. Combined with a growing sensitivity to detect even further diverged RdRP (Charon et al., 2022) it is reasonable to anticipate and begin engineering data-solutions which will scale for the interpretation of the anticipated 100 million distinct viruses which will be sequenced by the end of the decade. Human curation of viruses is already overtaxed and thus machine-augmented interpretation of viruses will be a central problem of computational virology in the coming years.
Establishment of similar gene-markers for DNA viruses, retroviruses, ribozyme-viruses and bacteriophages will undoubtedly be achieved in the next couple of years and palmID can be extended to encompass such viruses. A critical limitation of this and all virus-screens and meta-data aggregation is that the presence of nucleic acids in a sample does not prove infection has occurred. Viral nucleic acids are highly abundant in our natural world, in common laboratory reagents (Asplund et al., 2019, Porter et al., 2021), and as cross-sequencing lane contaminants (barcode hopping). In addition, human meta-data annotation is notoriously error-prone, thus any virus-sample or virus-environment associations should be treated as hypotheses upon which further investigation is done.
palmID is a community project to achieve a meta-curation of known and novel RNA viruses as a proof-of-concept. The project is under active development and I am soliciting for community feedback, developer-, or virological collaborators for public-domain/open-source development.
If you have any comments, feature requests or suggestions please reach out directly or leave a comment.
*References*
1. Asplund M, Kjartansdóttir KR, Mollerup S, Vinner L, Fridholm H, Herrera JAR, et al. Contaminating viral sequences in high-throughput sequencing viromics: a linkage study of 700 sequencing libraries. Clinical Microbiology and Infection. 2019 Oct 1;25(10):1277–85.
2. Babaian A, Edgar RC. Ribovirus classification by a polymerase barcode sequence [Internet]. bioRxiv; 2021 [cited 2022 Apr 12]. p. 2021.03.02.433648. Available from: https://www.biorxiv.org/content/10.1101/2021.03.02.433648v1
3. Bennett AJ, Paskey AC, Ebinger A, Pfaff F, Priemer G, Höper D, et al. Relatives of rubella virus in diverse mammals. Nature. 2020 Oct;586(7829):424–8.
4. Buchfink B, Reuter K, Drost H-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods. 2021 Apr;18(4):366–8.
5. Charon J, Buchmann JP, Sadiq S, Holmes EC. RdRp-scan: A Bioinformatic Resource to Identify and Annotate Divergent RNA Viruses in Metagenomic Sequence Data [Internet]. bioRxiv; 2022 [cited 2022 Apr 12]. p. 2022.02.28.482397. Available from: https://www.biorxiv.org/content/10.1101/2022.02.28.482397v1
6. Cooper LZ. The history and medical consequences of rubella. Rev Infect Dis. 1985 Apr;7 Suppl 1:S2-10.
7. Edgar RC, Taylor J, Lin V, Altman T, Barbera P, Meleshko D, et al. Petabase-scale sequence alignment catalyses viral discovery. Nature. 2022 Feb;602(7895):142–7.
8. Ellison A, Zamudio K, Lips K, Muletz-Wolz C. Temperature-mediated shifts in salamander transcriptomic responses to the amphibian-killing fungus. Molecular Ecology. 2020;29(2):325–43.
9. Grimwood RM, Holmes EC, Geoghegan JL. A Novel Rubi-Like Virus in the Pacific Electric Ray (Tetronarce californica) Reveals the Complex Evolutionary History of the Matonaviridae. Viruses. 2021 Mar 31;13(4):585.
10. Katz KS, Shutov O, Lapoint R, Kimelman M, Brister JR, O’Sullivan C. STAT: a fast, scalable, MinHash-based k-mer tool to assess Sequence Read Archive next-generation sequence submissions. Genome Biol. 2021 Sep 20;22(1):270.
11. Neri U, Wolf YI, Roux S, Camargo AP, Lee B, Kazlauskas D, et al. A five-fold expansion of the global RNA virome reveals multiple new clades of RNA bacteriophages [Internet]. bioRxiv; 2022 [cited 2022 Apr 12]. p. 2022.02.15.480533. Available from: https://www.biorxiv.org/content/10.1101/2022.02.15.480533v2
12. Porter AF, Cobbin J, Li C-X, Eden J-S, Holmes EC. Metagenomic Identification of Viral Sequences in Laboratory Reagents. Viruses. 2021 Oct 21;13(11):2122.
13. Wolf YI, Kazlauskas D, Iranzo J, Lucía-Sanz A, Kuhn JH, Krupovic M, et al. Origins and Evolution of the Global RNA Virome. mBio. 9(6):e02329-18.
14. Wu Z, Yang L, Ren X, He G, Zhang J, Yang J, et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 2016 Mar;10(3):609–20.
15. Xia Y, Luo W, Yuan S, Zheng Y, Zeng X. Microsatellite development from genome skimming and transcriptome sequencing: comparison of strategies and lessons from frog species. BMC Genomics [Internet]. 2018 [cited 2022 Mar 14];19. Available from: https://www.ncbi.nlm.nih.gov/labs/pmc/articles/PMC6286531/
16. Zayed AA, Wainaina JM, Dominguez-Huerta G, Pelletier E, Guo J, Mohssen M, et al. Cryptic and abundant marine viruses at the evolutionary origins of Earth’s RNA virome. Science. 2022 Apr 8;376(6589):156–62.