Note: This post is part of ongoing work and will be included in a forthcoming publication about the evolutionary processes of Ebola virus in reservoir hosts. We discuss this here because we believe it may be relevant to understanding the zoonotic source of the recently announced Ebola virus outbreak in Kasai Province of DRC.
At the start of the West African Ebola virus epidemic in 2014, when the first Guinea genomes were released [1], it was noticed that the new outbreak was relatively divergent to the known EBOV diversity. Initial rooting (using outgroup of other Ebolavirus species) gave the West African lineage as a sibling to Central African lineage.
Dudas and Rambaut [2] suggested that a simple molecular clock dating would support the 1970s genomes as the root with the implication that all the human outbreaks including the West African derive from very limited diversity (a bottle-neck?) in the 1970s with continent-wide dispersal since then. Such a rooting was supported by a compelling root to tip divergence plot which suggested an evolutionary rate similar to that observed in human outbreaks. This same rooting and rate were recovered by a relaxed clock BEAST analysis.
However every outbreak since the West African outbreak of 2013 has fallen short of the divergence expected under this model, leading some to suggest that EBOV has entered a different reservoir [3]. The most recent outbreak, in Kasai Province, August 2025, continues this trend (https://virological.org/t/1003). Despite emerging nearly 50 years after the first outbreaks were observed, genetic sequencing has revealed this strain is closely related to those sampled in the 1970s. Does this outbreak also represent a spill over from a new reservoir?
The EBOV phylogeny is split into several clades of geographically clustered outbreaks. We noticed that previous root-to-tip correlations relied heavily on the long branches connecting clusters, and that this trend differs from that observed within clusters. We searched for a possible rooting that would provide similar within- and between- cluster correlations.
There is no rooting that does not require outliers with less divergence than their date would suggest. However, if we root the Ebola phylogeny as in Figure 1B, it is possible to find a subset of branches that pass through the root and connect several outbreak clusters with a similar between- and within- cluster evolutionary rate. This model suggests EBOV diversity is older than previously anticipated with an estimated root date in the 1950s, and the reservoir contained significant diversity prior to the 1970s outbreaks. It also implies outliers were already present before 2013 (DRC 1995 and DRC 2007/8). Unaccounted for rate heterogeneity would explain why the 1970s root is not recovered with a priori local clocks placed on other branches [4, 5].
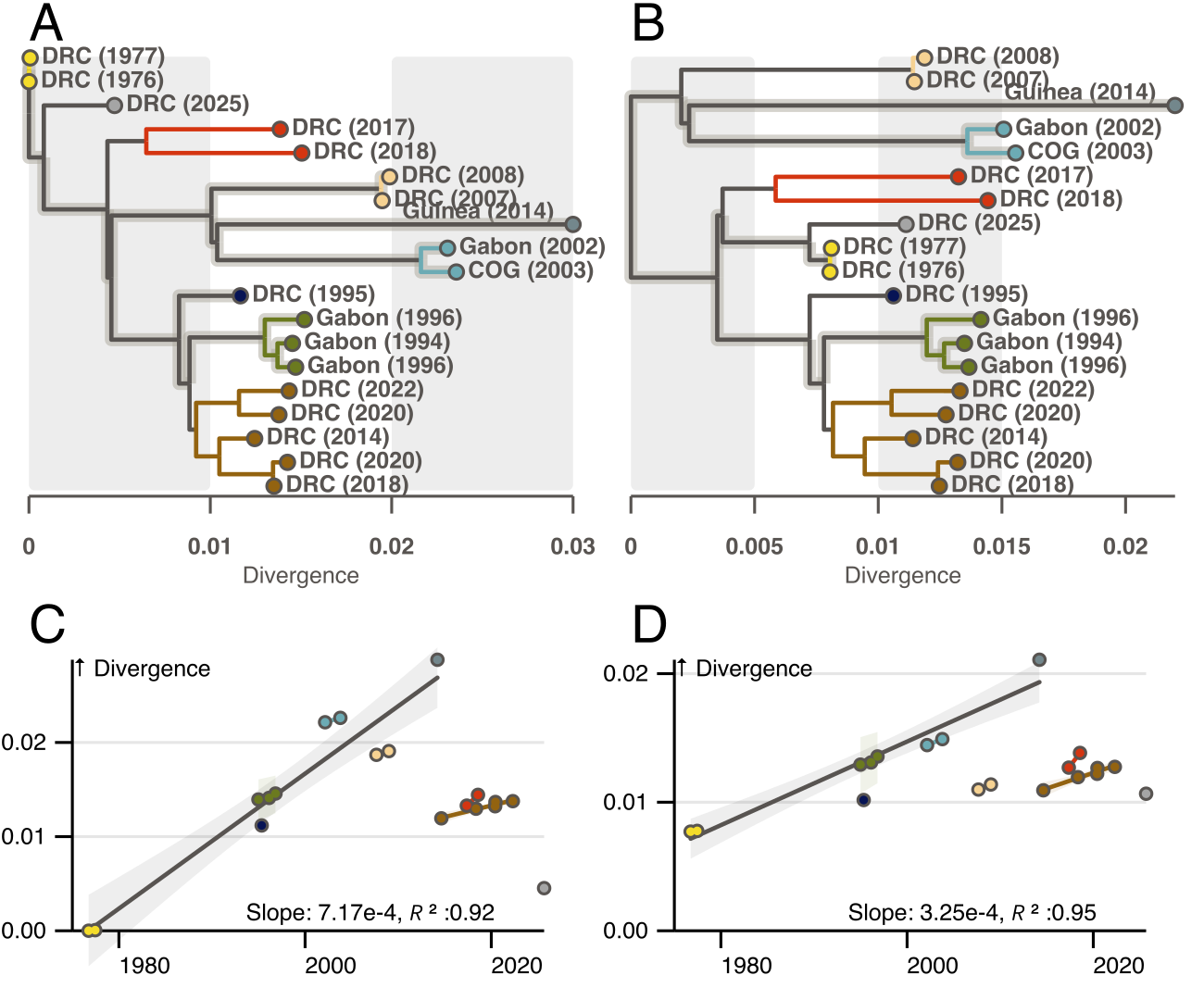
Figure 1 | Root to tip exploration of rate heterogeneity in the EBOV phylogeny. A and C) The EBOV phylogeny rooted at the accepted 1970s outbreak position and corresponding root to tip divergence plot. B and D) Similar to A and C but with the novel proposed rooting. In all figures tips are colored by outbreak cluster and labeled by country and year of outbreak. Solid Branches in A and B connect tips used in the regressions in C and D. Unhighlighted black branches subtend clades not included in the between cluster regression, and are hypothesized locations for evolutionary slow-down events.Smaller colored regressions connect tips belonging to the same clusters.
In light of this, we believe the recent outbreak is one of many outliers that deviate from an underlying evolutionary rate model according to a specific process. We hypothesize that the EBOV evolution alternates between two processes. The virus spreads through the reservoir population with an evolutionary rate of roughly 3x10-4 subst/site/year. This spread seeds clusters of zoonotic outbreaks. However, periodically, the virus undergoes delayed replication and slower rates of evolution. This process likely occurs on several branches in the Ebola phylogeny (dashed in figure 1) and results in clusters of outbreaks that are less diverged than expected given the standard model.
We suggest that this process is analogous to, or at least related to, the phenomenon observed in human outbreaks where the virus becomes quiescent in an individual after the initial acute infection. In a number of recorded cases the virus later proliferates again and results in an outbreak many months [6], or in one case years [7], after the initial infection.
Little is known regarding EBOV dynamics outside of human to human transmission, and our understanding of the viral reservoir is largely informed by what we can infer from the phylogeny connecting these outbreaks. We propose a new molecular clock informed rooting based on the assumption that large deviations in EBOV’s evolutionary rate are caused by periods of quiescence in the reservoir. The new sequences from the August 2025 outbreak in Kasai Province are consistent with this model and suggest the process is widespread. It seems plausible that the unknown process behind EBOV quiescence and reemergence is not unique to human infections.
Authors and affiliations
John T. McCrone1, Guy Baele2, Luiz M. Carvalho3, Gytis Dudas4, Ifeanyi Omah5, Eddy Kinganda-Lusamaki6, 7, Placide Mbala-Kingebeni6, 8, Marc A. Suchard9, 10, 11 and Andrew Rambaut5
- Vaccine and Infectious Disease Division, Fred Hutchinson Cancer Center, Seattle, Washington, USA
- Department of Microbiology, Immunology and Transplantation, Rega Institute, KU Leuven, Leuven, Belgium
- School of Applied Mathematics, Getulio Vargas Foundation (FGV), Rio de Janeiro, Brazil
- Institute of Biotechnology, Life Sciences Centre, Vilnius University, Vilnius, Lithuania
- Institute of Ecology and Evolution, University of Edinburgh, Edinburgh, UK
- INRB, University of Kinshasa, Kinshasa, DRC
- TransVIHMI, Université de Montpellier, INSERM, IRD, Montpellier, France
- South African National Bioinformatics Institute, University of the Western Cape, South Africa
- Department of Biostatistics, Fielding School of Public Health, University of California, Los Angeles, CA, USA
- Department of Biomathematics, David Geffen School of Medicine, University of California, Los Angeles, CA, USA
- Department of Human Genetics, David Geffen School of Medicine, University of California, Los Angeles, CA, USA
References
- Baize S, Pannetier D, Oestereich L, Rieger T, Koivogui L, Magassouba N ’faly, et al. Emergence of Zaire Ebola Virus Disease in Guinea. N Engl J Med. 2014;371: 1418–1425.
- Dudas G, Rambaut A. Phylogenetic Analysis of Guinea 2014 EBOV Ebolavirus Outbreak. PLoS Curr. 2014;6. PMC4024086
- Lam TT-Y, Zhu H, Chong YL, Holmes EC, Guan Y. Puzzling origins of the Ebola outbreak in the Democratic Republic of the Congo, 2014. J Virol. 2015; JVI.01226–15.
- Ebola Virus Local Clock Analysis | BEAST Documentation
- Mbala-Kingebeni P, Aziza A, Di Paola N, Wiley MR, Makiala-Mandanda S, Caviness K, et al. Medical countermeasures during the 2018 Ebola virus disease outbreak in the North Kivu and Ituri Provinces of the Democratic Republic of the Congo: a rapid genomic assessment. Lancet Infect Dis. 2019;19: 648–657.
- Diallo B, Sissoko D, Loman NJ, Bah HA, Bah H, Worrell MC, et al. Resurgence of Ebola Virus Disease in Guinea Linked to a Survivor With Virus Persistence in Seminal Fluid for More Than 500 Days. Clin Infect Dis. 2016;63: 1353–1356.
- Keita AK, Koundouno FR, Faye M, Düx A, Hinzmann J, Diallo H, et al. Resurgence of Ebola virus in 2021 in Guinea suggests a new paradigm for outbreaks. Nature. 2021;597: 539–543.