Omicron is a Multiply Recombinant Set of Variants That Have Evolved Over Many Months
William R. Gallaher
Mockingbird Nature Research Group, Pearl River, LA 70452
and Dept. of Microbiology, Immunology and Parasitology, Louisiana State University Health, 1901 Perdido St., New Orleans LA 70112
Over the last several weeks, a novel variant of concern (VOC) of SARS-CoV-2, designated B.1.1.569, has appeared in South Africa and is rapidly spreading around the world. Within the last week or so, two sub-variant species have been identified, named BA.1 and BA.2. While it has already been noted that the two sub-variants are recombinants, it is not generally appreciated how multiply recombinant they are, nor is it yet clear from what progenitor Omicron arose, or over what time scale.
From the list of non-synonymous mutations (see Excel panel below) it is possible to delineate as many as 8, and at the very least 4 different segments that differ in their relatedness between BA.1 and BA.2, as well as their relatedness to a pre-Alpha sequence, dating to early 2020, from which most of the Omicron sequence appears to be derived.
| VARIANT | ||||
|---|---|---|---|---|
| OMICRON | ||||
| B.1.1.7 (Alpha) | BA.1 | BA.2 | ||
| nsp1 S135R | ||||
| PLpro F24I | ||||
| PLpro K38R | ||||
| PLpro T183I | ||||
| PLpro G489S | ||||
| PLpro A890D | ||||
| PLpro S1265I | ||||
| PLpro del1266 | ||||
| PLpro I1412T | ||||
| Plpro A1892T | ||||
| nsp4 L264F | ||||
| nsp4 T327I | ||||
| nsp4 L438F | ||||
| nsp4 T492I | nsp4 T492I | |||
| 3CL P132H | 3CL P132H | |||
| nsp6 L105F | ||||
| nsp6 S106K | ||||
| nsp6 del107-109 | nsp6 del106-108 | nsp6 del106-108 | ||
| nsp6 F109L | ||||
| nsp6 I189L | ||||
| RDRP P323L | RDRP P323L | RDRP P323L | ||
| nsp13 R392C | ||||
| nsp14 I42V | nsp14 I42V | |||
| nsp15 T112I | ||||
| S T19I | ||||
| S L24S | ||||
| S 25PPAins | ||||
| S A67V | ||||
| S del69-70 | S del69-70 | |||
| S T95I | ||||
| S G142D | S G142D | |||
| S del144-145 | S del143-145 | |||
| S N211I | ||||
| S del212 | ||||
| S G213G | ||||
| S 215EPEins | ||||
| S G339D | S G339D | |||
| S S371L | S S371L | |||
| S S373P | S S373P | |||
| S S375F | S S375F | |||
| S T376A | ||||
| S D405N | ||||
| S R408S | ||||
| N440K | ||||
| S G446S | ||||
| S S447N | S S447N | |||
| S T478K | S T478K | |||
| S E484K | S E484A | S E484A | ||
| S Q493A | S Q493A | |||
| S Q498R | S Q498R | |||
| S N501Y | S N501Y | S N501Y | ||
| S Y505H | S Y505H | |||
| S T547K | ||||
| S A570D | ||||
| S D614G | S D614G | S D614G | ||
| S H655Y | S H655Y | |||
| S N679K | S N679K | |||
| S P681H | S P681H | S P681H | ||
| S T716I | ||||
| S N764K | S N764K | |||
| S D796Y | S D796Y | |||
| S Q954H | S Q954H | |||
| S N969K | S N969K | |||
| S L981F | ||||
| S S482A | ||||
| S D1118H | ||||
| Orf3a T223I | ||||
| E T9I | E T9I | |||
| M D3G | ||||
| M Q19E | M Q19E | |||
| M A63T | M A63T | |||
| Orf6 D61L | ||||
| Orf8 Q27* | ||||
| Orf8 R52I | ||||
| Orf8 Y73C | ||||
| N D3L | ||||
| N P13L | N P13L | |||
| N del31-33 | N del31-33 | |||
| N R203K | N R203K | N R203K | ||
| N G204R | N G204R | N G204R | ||
| N S235F | ||||
| N S413R |
Phylograms presented in preprint form (1) depict the origin of Omicron independently of any other variant, with an origin close to the common early branchpoint with Alpha and Lambda in the PGII group of variants. This variant has been evolving undetected for many months, producing not only a large of number of mutations, but a substantial variety between the two sub-species as well. Relative to the reference sequence designated by NCBI as NC_045512, there are 21 amino acid changes in Alpha (B.1.17 plus E484K), 49 in BA.1 and 51 in BA.2. I derived the above list list from the Stanford Variant graphic (2). Eight common substitutions are found throughout the genome between Alpha and BA.1, and six in BA.2. These provide a general Alpha-related roadmap to Omicron. They include the 3 amino acid deletion on nsp6, the RDRP P323L, the S del69-70 in BA.1, the S del144-145 in BA.1, the S E484K/A, S N501Y, S D614G, SP681H, and the trinucleotide substitution that yields N R203K and N G204K (the real purpose of which is to duplicate the ACGAAC splice acceptor site and a novel mRNA for the N2 region of N (3). These markers are sufficient to establish an Alpha like progenitor, even though the Alpha and Omicron lineages have a preponderant number of unique mutations indicating divergent evolution since early 2020.
The eight independent segments may be delineated as follows.
SEGMENT A
From Origin or nsp1 S135R (nt1 or 640)
To nsp14 L438F (nt9868)
then crosspoint 1
SEGMENT B
From nsp4 T492I (nt10030)
To nsp14 I42V (nt18164)
then crosspoint 2
SEGMENT C
From nsp15 T112I (nt19956)
To S211-215 hotspot (nt22258)
then crosspoint 3
SEGMENT D
From S G339D (nt22580)
To S S375F (nt22688)
then crosspoint 4 ?
SEGMENT E
From S T376A (nt22689)
To S G446S (nt22901)
then crosspoint 5 ?
SEGMENT F
From S S447N (nt22902)
To S N969K (nt24470)
then crosspoint 6 ?
SEGMENT G
From S L981F (nt24506)
To Orf3a T223I (nt26061)
then crosspoint 7 ?
SEGMENT H
From E T9I (nt26271)
To END (nt29903)
Segment A runs either from the Origin or from nsp1 S135R at nt 640, to nsp4 L438F at nt 9868. In this segment, Alpha has 3 amino acid substitutions, BA.1 has 4, and BA.2 has 6, and none of them correspond to one another. Enumerating synonymous mutations may shed more light, but clearly the first third of the genome seems to be derived from three independent sources. Importantly BA.1 and BA.2 are clearly nonidentical and recombinant relative to one another over this expanse. Phylogenetic analysis that homogenizes such a large recombinant segment with the whole genome introduces a serious error into the phylogenetic tree. True trees exclude recombinant sequence from consideration in assessing evolutionary relatedness.
Segment B has a high degree of commonality, in sharp contrast to segment A. It begins with nsp4 T492I, at nt 10030 and appears to end at nsp14 I42V at 18164. Alpha has 3 substitutions in this segment, BA.1 has 7 and BA.2 has 8. Five of the 8 substitutions are shared between BA.1 and BA.2, indicating relatedness with divergence. Two of these 5 are also shared with Alpha. This segment can be reasonably judged as non-recombinant across the three variant sequences.
Segment C begins with nsp15 T112I and may end at the S 211-215 hotspot we designated Indel Region 4 (IR4) elsewhere (4). In this segment there are 2 substitutions in Alpha, 8 in BA.1 and 6 in BA.2. Only 1 is shared between BA.1 and BA.2. Two of the 3 in Alpha are shared with BA.1, indicating that it is likely this segment in BA.2 is recombinant from an unknown source, while BA.1 in this segment is more likely divergent from a pre-Alpha progenitor. Four of the Omicron substitutions are between 211 and 215, possibly indicating variation in recombination. The S 215EPE insert is located at the end of this segment, reinforcing the hypothesis that variable copy-choice outcomes, at the carboxy-terminal margin of the recombination, may be responsible for the variation seen here. The result of the disparity here is that BA.1 and BA.2 have quite different NTD regions of the S glycoprotein, from apparently different sources.
Segment D is marked by an abrupt commonality between BA.1 and BA.2. It begins at S G339D and ends at S S375 F. All four substitutions in BA.1 are shared by BA.2. There are no substitutions in Alpha in this segment for comparison. This may also be reasonably judged a non-recombinant region between BA.1 and BA.2 sub-variants of Omicron.
Segment E begins at S T376A and ends at S G446S. It may either be recombinant or simply divergent between BA.1 and BA.2. However, the 2 substitutions in BA.1 and 3 in BA.2 do not correspond at all with one another. There are no substitutions in Alpha for comparison. It is short, only 0.2 kb, which also argues against it being a recombinant segment.
Segment F begins with a run of commonality that abruptly begins with S S447N and runs for over 1.5 kb to at least S N969K, if not in fact to the end of the genome. Over this segment, there are six substitutions in Alpha, 16 in BA.1 and 15 in BA.2. Four of the 6 positions in Alpha are also mutated in BA.1 and BA.2, convincingly aligning the three sequences. Fifteen substitutions in BA.1 are identical in BA.2. This is clearly a non-recombinant region of the genome, marked by common divergence of the two Omicron sub-variants from a pre-Alpha progenitor.
Segment G, from S L981F to Orf3a T223I, slightly interrupts this commonality, with a substitution in BA.1 and another in BA.2 that are non-identical with either one another or two others in Alpha. This may simply represent divergence, but is delineated because of the interruption, for 1.5 kb, in what was just previously virtually identical.
Segment H picks up the commonality again, from E T9I through the end of the genome. There are 8 substitutions in BA.1 and 9 in BA.2. Seven of these are identical, indicating a non-recombinant region for the last 3.6 kb of the genome.
If segment G is merely divergent, then this carboxy-terminal non-recombinant region may extend back to nt 22902, a full 7.0 kb. If what was delineated as segment E is likewise merely divergent, then the last 7.4 kb may be non-recombinant between BA.1 and BA.2. However, if that were true, then that extends the time of divergence between the two sub-variants in order to account for the number of different amino acid substitutions observed.
This would still leave separate segments, A, B, C, and D through the end of the genome, that cannot be reasonably accounted for except by multiple recombination events, with A and C the recombinant, likely in BA.2, and B and D the non-recombinant but divergent segments.
Graphically, the segments may be illustrated on the 30 kb genome, with at least 12.7 kb, or 42% of the genomes as recombinant from unidentified sources, as follows:
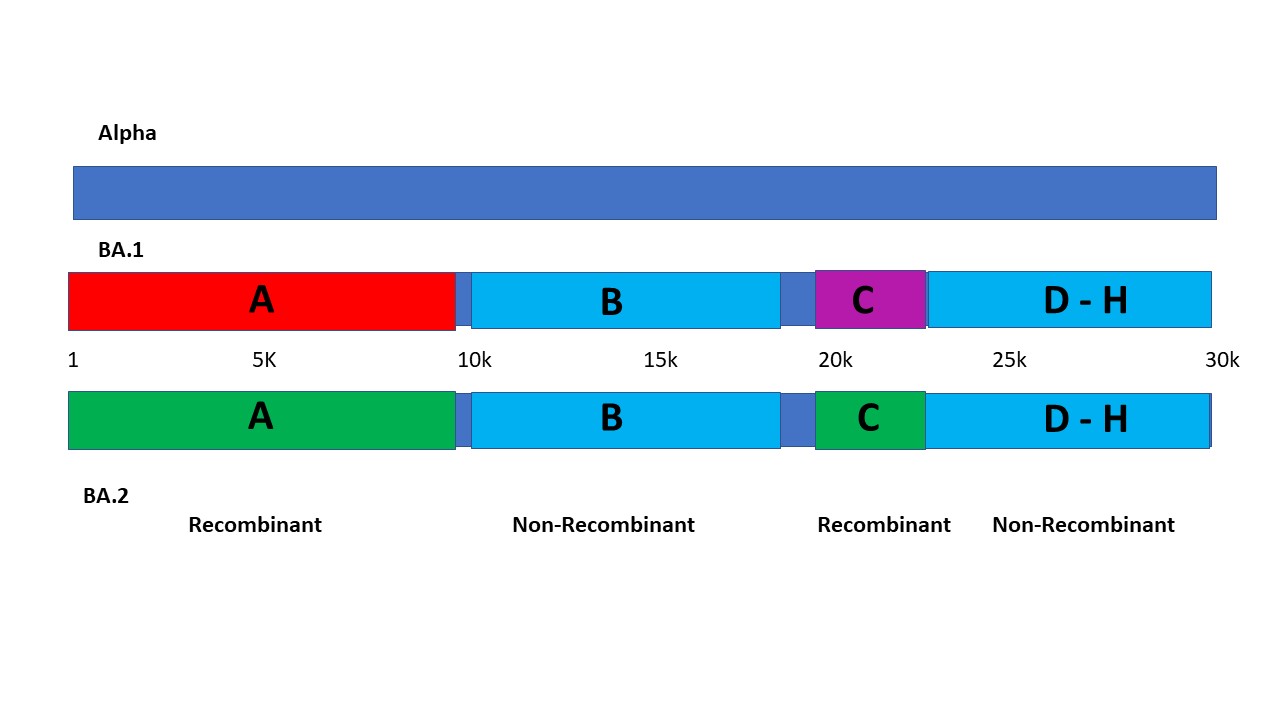
If Segments E and G are also recombinant, rather than merely divergent, this would add an additional 1.7 kb, raising the total to 14.4kb of recombinant sequence, or roughly 50% of the whole genome.
Overall, BA.1 and BA.2 only share 30 of their 49 and 51 amino acid substitutions. The total number of substitutions and the divergence between the two, even allowing for multiple recombination events, takes a good deal of time, likely extending back well into 2020. Where and how this occurred without discovery for all that time is a serious issue. Unidentified sources for a substantial stretch of sequence clearly indicate that there is a pool of SARS-CoV-2 variant sequences that remain undetected.
The discomfort in discovering an entirely new and widely divergent VOC, with two very different recombinant forms, is very real. Beyond the medical impact of the Omicron variants, there is every reason to believe that this will happen yet again, as it did for Delta and now with Omicron. The prospect of a widely divergent, actively recombining, and highly novel additional variant replicating, mutating and recombining completely under our radar is a chilling thought. One thing is certain: unless we can bring the global rate of replication of SARS-CoV-2 down by orders of magnitude, the old adage about the possible becoming probable and the probable becoming inevitable is bound to come true.
I am, and always have been, a strong proponent of vaccines. My wife and I have been fully immunized and boosted as soon as humanly possible. Still, I feel compelled to warn that we may not be able to immunize our way out of this, as immune escape becomes increasingly prioritized as an evolutionary pressure in the generation of variants. We need to be careful about being addicted to our high tech solutions, that meet considerable resistance and global supply chain and delivery issues. As some countries have discovered, viral epidemiology 101, interrupting the chain of infection, is vital. Masking, handwashing, sanitizing and social distancing – keeping infected folks from uninfected folks – remains the surest path to reducing the reproduction number anywhere. Cheap. low tech and effective means can be most broadly applied globally.
In the United States, we are on track to have 1,000,000 Americans dead from COVID, within basically two years of onset of the pandemic here. What was inconceivable here has become almost inevitable. That we are not doing every conceivable thing to stop this carnage here and globally is beyond unacceptable.
Acknowledgements:
The author has no institutional or extramural support, but also therefore no conflicts.
I would like to thank Bob Garry of Tulane, Steve Shafer of Stanford, and Brian Foley of the Los Alamos National Laboratory for helpful remarks in the last month, but I remain solely responsible for the content of this post.
References:
- Bansal K and Kumar S. 2021. Mutational cascade of SARS-CoV-2 leading to evolution and emergence of omicron variant. doi: https://doi.org/10.1101/2021.12.06.471389.
- Stanford Coronavirus Antiviral & Resistance Database (CoVDB)
- https://pando.tools/t/genomic-gymnastics-in-the-nucleocapsid-gene-of-sars-cov-2-during-transmission-in-humans-transposition-of-acgaac-and-creation-of-the-novel-n2-gene/715/5’
- https://pando.tools/t/spike-protein-mutations-in-novel-sars-cov-2-variants-of-concern-commonly-occur-in-or-near-indels/605.