The ongoing mpox virus outbreak of Kamituga in South Kivu province, Democratic Republic of Congo, is associated with a novel Clade I sublineage
Authors:
Leandre Murhula Masirika1,2, Jean Claude Udahemuka3,4, Leonard Schuele5, Pacifique Ndishimye4,6,7, Saria Otani8, Justin Bengehya Mbiribindi9, Jean M. Marekani10, Léandre Mutimbwa Mambo11, Marjan Boter5, David F. Nieuwenhuijse5, Trudie Lang12 ,Bilembo Kitwanda Steeven9, Nadine Malyamungu Bubala9, Ernest Balyahamwabo Kalalizi2, Jean Pierre Musabyimana4, Frank M. Aarestrup8, Marion Koopmans5, Bas B. Oude Munnink5*, Freddy Belesi Siangoli13*
Affiliations:
1 Centre de Recherche en Sciences Naturelles de Lwiro, Sud -Kivu, DS Bukavu, Democratic Republic of Congo (DRC)
2 SaBio Instituto de Investigación en Recursos Cinegéticos IREC (Universidad de Castilla-La Mancha & CSIC), Ronda de Toledo 12, Ciudad Real, Spain
3 Department of Veterinary Medicine, University of Rwanda, P.O. Box 57, Nyagatare, Rwanda
4 Stansile Research Organization, Kigali, Rwanda
5 Department of Viroscience, Erasmus University Medical Center, Rotterdam, The Netherlands
6 Department of Microbiology and Immunology, Dalhousie University, Halifax, Nova Scotia, Canada
7 African Institute for Mathematical Sciences, Kigali, Rwanda
8 Research Group for Genomic Epidemiology, National Food Institute, Technical University of Denmark, 2800 Kgs. Lyngby, Denmark
9 Hopital General de Reference de Kamituga, Sud- Kivu, DRC
10 Unit of Animal Production and Health, Nature Conservation and Development, Department of Biology, Faculty of Science, University of Kinshasa, P.O.box 218 Kinshasa 11 DRC
11 Zone de Santé de Kamituga, Sud- Kivu, DRC
12 The Global Health Network, Oxford University, UK
13 Division Provinciale de la Santé, Sud- Kivu, RDC
- These authors contributed equally
Introduction
Mpox virus (MPXV) is an emerging zoonotic disease belonging to the Poxviridae family and the genus Orthopoxvirus. Sporadic outbreaks of MPXV with increased frequency have been described in the Democratic Republic of the Congo (DRC) since the first detection in 1970 (Ladnyj et al, 1972). However, in 2022, a multi-country global outbreak of MPXV (Clade IIb) was declared by the WHO. The outbreak was predominantly described in Men Having Sex with Men (MSM) communities, although infections were also reported in individuals engaged in sexual contact with infected individuals.
Since the beginning of 2023 there have been reports of a sharp increase of suspected MPXV cases in the Democratic Republic of Congo (DRC) with the highest annual number of cases ever reported, new cases in geographic areas that had previously not reported mpox and an estimated case fatality rate of 4.6% (https://www.who.int/emergencies/disease-outbreak-news/item/2023-DON493). Despite the seeming severity of the situation only very basic genomic information is currently available, suggesting the DRC outbreak is caused by Clade I viruses and is sexually transmissible (Kibungu et al, 2024). However, in view of the ongoing outbreak and suggestions of changing epidemiology, obtaining sequence information of the outbreak strain(s) is crucial. This sequence information is also essential to evaluate if this novel strain can still be detected by the current molecular assays used to diagnose MPXV infections. Therefore, MPXV whole genome Nanopore sequencing training was given in Rwanda to train local scientists from Rwanda and DRC to perform both wet-lab sequencing and dry-lab sequence analysis. Here, we present the genome sequences from recently diagnosed cases in South Kivu, DRC.
Material and Methods
Routine sampling was done for the hospitalised patients, afterwards samples were sent to INRB Goma for PCR confirmation. Skin lesions and oral pharyngeal swabs were collected from 10 patients hospitalised for mpox in the Kamituga hospital, located in Kamituga health zone. In this region the first cases were reported from September 2023 onwards. Patients were between 17-26 years old and 5 were males and 5 females. Regarding the profession, 1 person was a driver , 1 person was working in the mines and 8 persons were sex workers.
The samples were collected in the first month of January 2024. Samples were stored in virus transport medium and frozen at -20°C. DNA was extracted with the Blood & Tissue Kit (Qiagen) according to the manufacturer’s recommendations. Viral transport medium was used as a negative control. Mpox DNA was amplified using a Mpox amplification scheme specifically designed for Mpox Clade IIb viruses to generate 2,500 bp amplicons and quantified using the Qubit dsHS DNA assay (ThermoFisher) (Welkers et al, 2022). Sequencing libraries were prepared using the Native Barcoding Kit 24 V14 (SQK-NBD114.24) (Oxford Nanopore Technologies).
Sequencing reads were basecalled using dorado v.7.2.13 with the super-accurate base calling model (ONT). Primer sequences were removed using cutadapt v4.6 (GitHub - marcelm/cutadapt: Cutadapt removes adapter sequences from sequencing reads) and aligned against a MXPV reference (GenBank accession: OQ729808) in MinKNOW v23.11.5 (ONT). The consensus was created from the resulting bam file using samtools v1.19.2 as described (Schuele et al., 2024; Nieuwenhuijse et al, 2022). NextClade v3.1.0 was used for clade assignment and quality checks (https://clades.nextstrain.org/). Sequences were aligned using MAFFT v7.520 (MAFFT - a multiple sequence alignment program), phylogenetic analysis was performed using IQ-TREE v2.2.6 (GitHub - iqtree/iqtree2: NEW location of IQ-TREE software for efficient phylogenomic software by maximum likelihood http://www.iqtree.org) and visualised with FigTree v1.4.4 (FigTree). In addition, phylogenetic analysis was performed using NextStrain CLI v8.2.0 and visualised in auspice (GitHub - nextstrain/cli: The Nextstrain command-line interface (CLI)—a program called nextstrain—which aims to provide a consistent way to run and visualize pathogen builds and access Nextstrain components like Augur and Auspice across computing environments such as Docker, Conda, and AWS Batch.). Consensus sequences were uploaded on GISAID (EPI_ISL_18886301, EPI_ISL_18886588, EPI_ISL_18886467, EPI_ISL_18886467, EPI_ISL_18886588 and EPI_ISL_18886301). Raw sequence data was uploaded to the ENA (accession IDs pending).
Results
At the time of writing, no mpox near-complete sequences circulating in Congo from the 2023 outbreak were available on (public) online databases. In this study, we applied targeted amplicon sequencing to characterise mpox sequences in patients visiting a hospital in Kamituga, DRC. Near-complete MPXV sequences were generated from 6/10 patients and all were classified as Clade I. The genome coverage ranged between 93.5% - 100% (average of 95.2%) (Supplementary Table 1 and 2).
Phylogenetic analysis was done including 94 complete genome reference mpox sequences from Africa available from GISAID on the 8th of February 2024 and two sequences from the recent 2022 global mpox outbreak. The new sequences clustered with currently published Clade I sequences, but distinct from all other Clade I sequences from DRC, suggesting the ongoing outbreak in South Kivu results from a separate introduction (Figure 1 and Supplementary Figure 1 and 2). The 6 sequences have several SNPs differences between the genomes which suggests ongoing circulation of this outbreak strain for some time already.
The newly sequenced sequences were aligned to closely related Clade I sequence EPI_ISL_13056243 and compared to the primer and probe sequences which are being recommended by the CDC to diagnose MPXV using an in house Primer Check Tool. While the generic primers still seem to be functional with only one mutation in the reverse primer, the specific Clade I virus target, recommended by the CDC, is deleted in the novel MPXV strain (Figure 2 and Supplementary Figure 3). Consequently, the rapid clade designation of newly diagnosed mpox cases outside of the African region is most likely not reliable for detection of this novel virus.
Discussion and Outlook
Phylogenetic analysis showed that the Kamituga, South Kivu, DRC mpox sequences are at the root of the Clade I viruses previously sequenced from the DRC suggesting a new introduction, most likely from a zoonotic reservoir. Although sequences from a small 2023 Kinshasa outbreak are not publicly available, the placement of those sequences in a published phylogenetic tree (Kibungu et al, 2024) suggests that the Kamituga outbreak is not related to the outbreak in Kinshasa. Our findings suggest that there are at least two independent outbreaks ongoing.
Multiple amplicon-based assays targeting Clade IIb MPXV have been developed and applied since the global Mpox outbreak in 2022 (Welkers et al, 2022; Brinkman et al, 2024). The application of a large amplicon assay which was originally designed to target Clade IIb resulted in an average genome coverage of 95.2% in the new Clade I genome, showing that this method can also be used to sequence Clade I viruses, although minor adjustments of primer pairs might result in more complete consensus sequences. Also, the consensus was generated using a reference based alignment using Nanopore data. Metagenomic Nanopore or Illumina sequencing might be needed in combination with de novo assembly approaches for refined consensus calling.
Remarkably, a large deletion in the genome of the novel MPXV was observed leading to failure of the clade I specific RT-PCR recommended by the CDC (Li et al, 2010). A deletion in the same region is also observed in clade II MPXV, which was the basis of the Clade assignment using the CDC PCR. Therefore, this rapid surveillance tool can no longer be used to rapidly detect Clade I virus infections in the ongoing global Clade IIb outbreak, if the viruses from the new lineage would be seeded internationally. Altogether, this data strongly suggests that whole genome sequencing and public data sharing of a larger subset of the current mpox cases in DRC is essential to understand the ongoing viral emergence and evolution. Further studies, sequencing and analyses are ongoing, but in accordance with the above statement we believe that rapid public sharing of all available information is essential to help to better understand and contain the current emergence of mpox.

Figure 1: Phylogenetic analysis of all whole genome mpox sequences from Africa available on GISAID including the 2024 sequences from Kamituga. Sequences from Kamituga are presented in Magenta, previous sequences from DRC in red and sequences from the 2022 outbreak in blue. Sequences with an arrow indicate sequences which clustered with the previous sequences from a 2023 MPXV outbreak from Kwilu Province near Kinshasa.

Figure 2: A) Single nucleotide mismatch with CDC recommended generic reverse primer (G2R_G reverse primer). B) Complete deletion of CDC recommended clade I specific forward, reverse, and probe target location (C3L). Visualised by primer-check tool: https://viroscience-emc.shinyapps.io/primer-check/.
Acknowledgements
This work was funded by the EU Horizon 2020 grants VEO (874735) and the Global Health EDCTP3 Joint Undertaking (Global Health EDCTP3) programme under grant agreement No. 101103059 (GREATLIFE).
We greatly thank Wildlife Conservation Network (WCN) and Conservation Action Research Network (CARN) for their support in terms of scholarship for the master’s research studies they awarded to the first author.
We greatly thank the University of Dalhousie of Canada for supporting the Kamituga hospital, and the Institut Nationale de Recherche Biomedicale (INRB) Goma for PCR diagnostics.
References
Ladnyj et al. “A human infection caused by monkeypox virus in Basankusu Territory, Democratic Republic of the Congo”. Bull World Health Organisation (1972).
Kibungu, Emile et al. “Clade I-associated mpox cases associated with sexual contact, the Democratic Republic of the Congo”. Emerging Infectious Diseases (2024)
Brinkmann, Annika, et al. “Genome sequencing of the mpox virus 2022 outbreak with amplicon-based Oxford Nanopore MinION sequencing.” Journal of Virological Methods (2024): 114888.
Nieuwenhuijse, David F., et al. “Towards reliable whole genome sequencing for outbreak preparedness and response.” BMC genomics 23.1 (2022): 1-9.
Schuele, Leonard, et al. “Circulation, viral diversity and genomic rearrangement in mpox virus in the Netherlands during the 2022 outbreak and beyond.” Journal of Medical Virology 96.1 (2024): e29397.
Welkers Matthijs Jonges, van den Ouden Anton 2022. Monkeypox virus whole genome sequencing using combination of NextGenPCR and Oxford Nanopore. protocols.io; Monkeypox virus whole genome sequencing using combination of NextGenPCR and Oxford Nanopore
Li et al. “Real-time PCR assays for the specific detection of monkeypox virus West African and Congo Basin strain DNA”. Journal of Virological Methods (2010)
Supplementary Files
Supplementary Table 1: Clade assignment and quality metrics of mpox consensus sequences.
| Sample ID | Cladea | Genome coverage | Coverage depth |
|---|---|---|---|
| 1 L | I | 100% | 6477.1x |
| 2 L | I | 93.5% | 959.2x |
| 3 L | I | 94.3% | 2420.6x |
| 4 L | I | 94% | 2950x |
| 7 O | I | 93.8% | 3186.3x |
| 9 L | I | 95.3% | 1709.9x |
a Clade assignment as defined by Nextclade v3.1.0. Abbreviations: L, Lesion (skin); O, oropharyngeal swab.
Supplementary Table 2: Clade assignment and sequence quality of 10 sampled patients with suspected mpox infection.
Patient ID Cladea Skin lesion swab Oropharyngeal swab
Genome coverage Coverage depth Genome coverage Coverage depth
1 I 100% 6477.1x 94.2% 2357.8x
2 I 93.5% 959.2x
3 I 94.3% 2420.6x
4 I 94% 2950x 86.2% 27.9x
5 I 90.9% 79x
6 I
7 I 93.5% 1232.3x 93.8% 3186.3x
8 I 94.2% 3214.5x
9 I 95.3% 1709.9x
10 I 91.4% 2866.2x
a Clade assignment as defined by Nextclade v3.1.0; Empty fields indicate no mpox amplicon product.
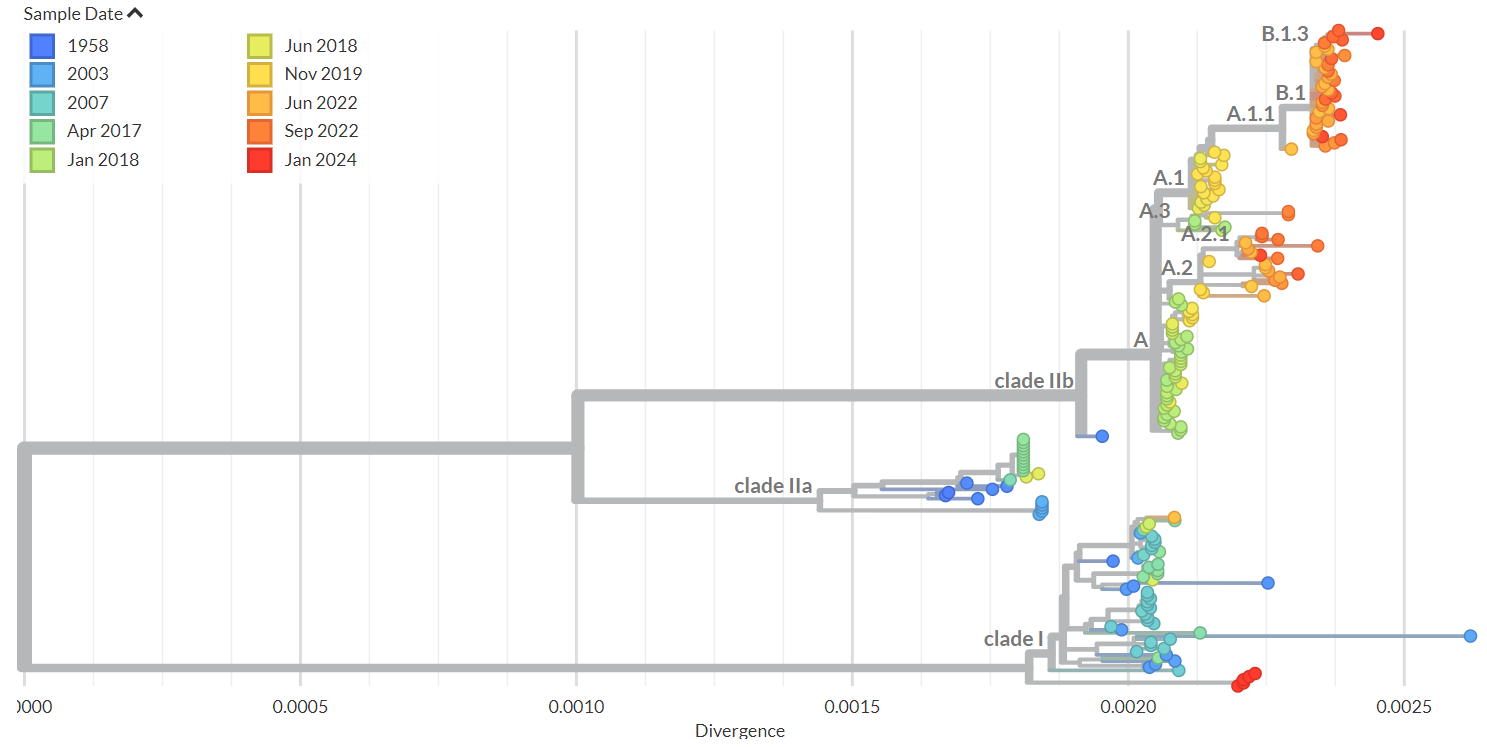
Supplementary Figure 1: Phylogenetic tree showing 211 mpox genomes including the six new sequences from Kamituga in dark red.
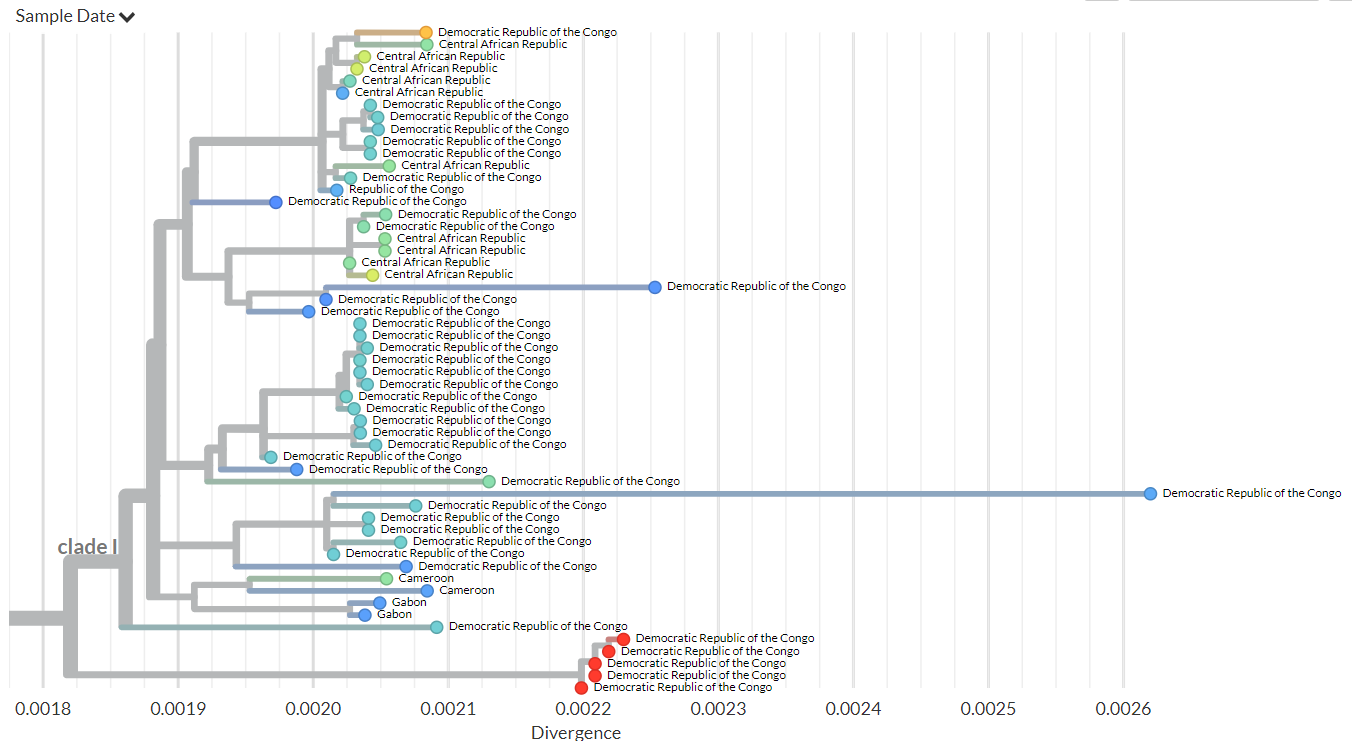
Supplementary Figure 2: Zoom of the phylogenetic tree focussing on the Clade I viruses. Sequences from this study are shown by enlarged red circles.
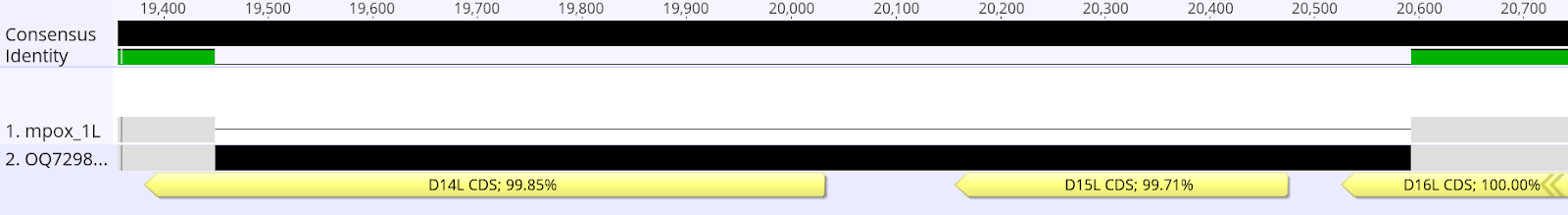
Supplementary Figure 3: Alignment of exemplary sequenced mpox sample and reference highlighting the deletion downstream of the 3’ ITR (position 19,451-20,593).