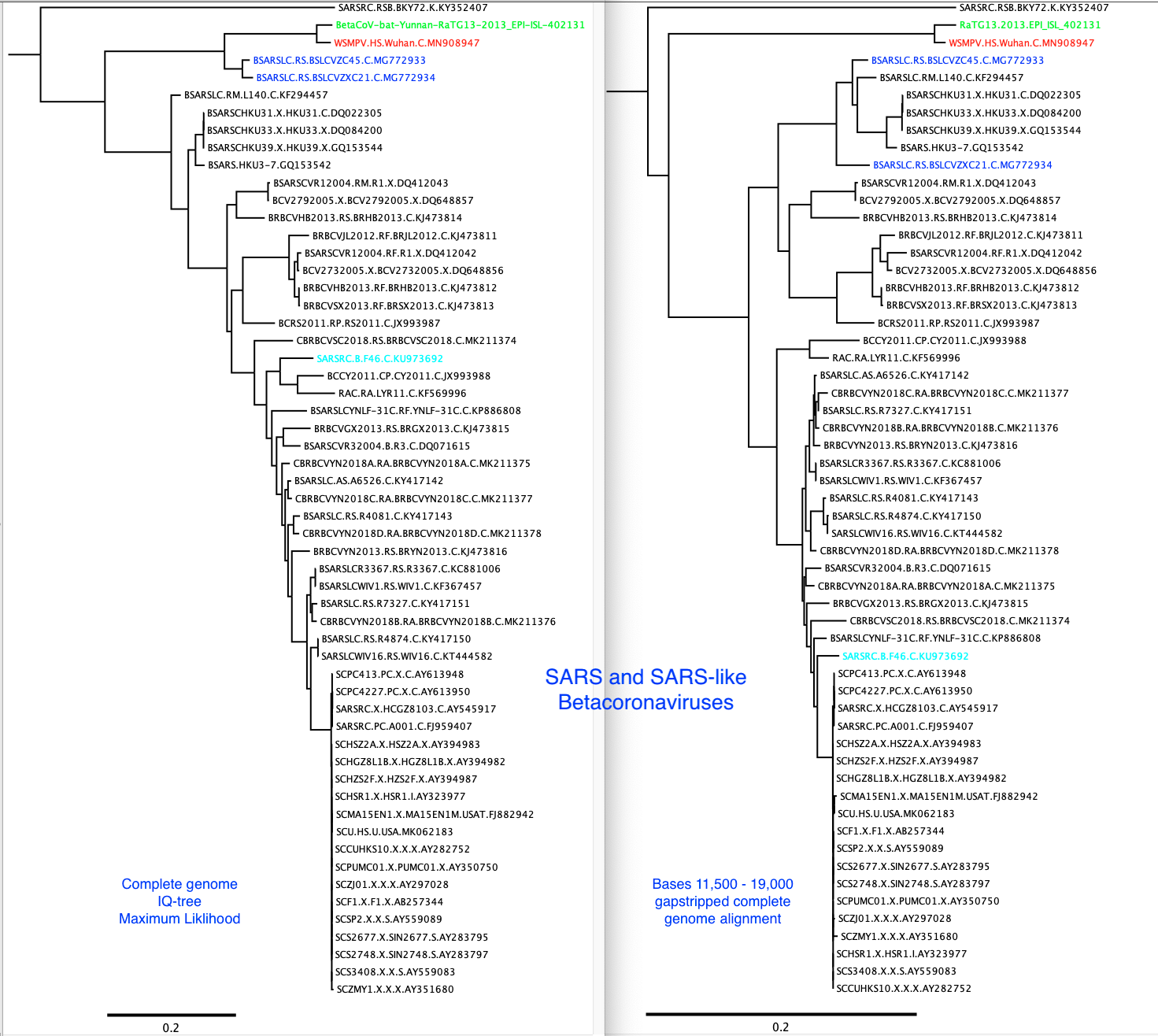
A 2013 bat Betacoronavirus (GISAID accession EPI_ISL_402131) was recently uploaded to GISAID. It is much more closely related to the 2019-2020 human outbreak viruses than the previously available bat virus genomes.
The publication of this sequence is available at the BioRxiv:
BetaCoronaviruses_56_WuhanCladePlus2013Bat_TreePDF.pdf (4.4 KB)

There is recombination between many of these viruses, so the phylogenetic trees built from complete genomes is not an accurate reflection of the evolutionary history of the various lineages.