Introduction
Case History:
On January 30, 2025, the Ministry of Health declared an outbreak of Sudan Ebola Virus (SUDV) Disease in Kampala, Uganda, following the death of a healthcare worker at Mulago National Referral Hospital. The index case, a 32-year-old male nurse, presented with fever and other symptoms on 19th January 2025 and sought care at three healthcare facilities, one in Kampala one in Wakiso and another in Mbale district, before succumbing to the illness 10 days later (29th January 2025) at the Mulago Supra-National Hospital (SNH). Cadaveric samples were collected on the same day, for ongoing Ebola Virus Disease (EVD) mortality surveillance and real-time PCR (RT-PCR) results from three independent laboratories confirmed Sudan ebolavirus (SUDV). Whole-genome sequencing was conducted on the same day with a turnaround time (TAT) from when the case was suspected, samples collected, results confirmed, sequencing performed, and report generation within 24 hours. Here, we present a preliminary whole genomic analysis undertaken at the Central Public Health Laboratories (CPHL) Core Genomics Facility using genetic material extracted from the index case sample (designated CL292200) referred to by the Central Emergency Response & Surveillance Laboratory.
History of SUDV Outbreaks in Africa:
First identified in 1976, SUDV is a highly pathogenic Ebola virus that causes severe hemorrhagic fevers and is responsible for multiple outbreaks in sub-Saharan Africa, specifically South Sudan, and Uganda (Figure 1). While the natural reservoir remains unknown, bats are still suspected of human transmission primarily occurring through contact with infected animals or their fluids, and direct contact with infected bodily fluids or contaminated surfaces. (Han et al., 2015; Leroy et al., 2005). SUDV is contagious and deadly, exhibiting a case fatality rate of about 53% (range: 41% to 100%) based on aggregated historical data. (Carbonnelle et al., 2022). Currently, there are no specific treatments or vaccines approved for SUDV. (World Health Organization, 2022).

Fig 1: A map of Africa showing countries (South Sudan and Uganda) that have ever reported an outbreak of Sudan Ebola virus disease since it was first identified in 1976.
Data before the current outbreak indicates that Uganda has experienced the second-highest number of Ebola virus disease (EVD) outbreaks in Africa overall, accounting for 16.6% (7/42), following the Democratic Republic of the Congo, which has recorded 35.7% (15/42). (Hussein, 2023). At a continental level, majority of these outbreaks have been caused by Zaire ebolavirus (ZEBOV: 73.8% [31/42]), followed by Sudan ebolavirus (SUDV: 19.0% [8/42]), Bundibugyo ebolavirus (BDBV: 4.8% [2/42]), and Taï Forest ebolavirus (TAFV: 2.4% [1/42]). (Hussein, 2023). On the contrary, considering the current outbreak, Uganda has so far had the highest number of SUDV outbreaks (5/8), with the last documented outbreak having occurred 2 years ago (September 2022-January 2023) as depicted in Map (Figure 1). In light of the just concluded 2022 SUDV outbreak in Uganda and heightened suspicions of resurgence, the Uganda Ministry of Health (MoH) tasked the laboratories to undertake genomic sequencing and phylogenetic analysis of the isolate responsible for the current 2025 outbreak.
Methods
RNA Extraction, Library Preparation, and Sequencing
A postmortem blood sample was collected and following virus inactivation with AVL buffer and carrier RNA from the Qiagen Viral RNA Mini Kit, RNA was extracted using a Qiagen QIAmp Viral RNA Mini Kit. Sequencing was then performed using amplicon-based and enrichment-based approaches on the Oxford Nanopore Technologies (ONT) GridION and Illumina MiSeq, respectively. For the amplicon-based method, artic-pan-ebola primer pools (version 1.0.0) (https://bit.ly/3EwAgYR), were used to generate amplicons which were then barcoded using the Rapid Barcoding Kit following a Midnight protocol. The resulting libraries were subsequently loaded on the GridION for real-time sequencing. Basecalling parameters were set to “Super Accuracy” to generate high-quality FASTQ files. Enrichment-based sequencing was performed using Illumina RNA Prep with enrichment using the Viral Surveillance Panel, generating libraries which were then loaded onto the MiSeq system.
Bioinformatics Data Analysis
For ONT analysis, FASTQ files were concatenated into one file per barcode which were then subjected to the ARTIC field bioinformatics pipeline (version 1.6.1) (fieldbioinformatics, 2018/2025) for variant calling and generation of consensus genomes for downstream phylogenetic analysis. The resultant consensus genome was concatenated with genomes from the 2022 Uganda outbreak and existing NCBI genomes of the SUDV. Multiple sequence alignment (MSA) was performed using the web version of MAFFT with default settings. (Katoh et al., 2019). The MSA was then used as input for IQ-TREE (version 2.3.6) for phylogenetic tree reconstruction. (Nguyen et al., 2015). To investigate the relationship between the current outbreak with previous in-country and neighbouring country outbreaks, a phylogenetic analysis was performed with selected sequences from 1976, 1979, 2000, 2004, 2011, and 2012, as well as the most recent outbreak in Uganda, 2022. (Nabadda et al., 2023). The generated phylogenetic tree was visualized and annotated using the Interactive Tree of Life (version 7) (Letunic & Bork, 2007) and rooted using MH121162.1, a sequence from the 1976 outbreak in Nzara, South Sudan.
For the Illumina data, FASTQ files were analyzed using a custom in-house pipeline. Briefly, the pipeline uses trim galore (version 0.6.10) with default settings to trim poor-quality reads. (Krueger, 2016/2025). The resultant good reads are mapped to the SUDV reference genome (accession NC_006432.1) using bwa (0.7.17-r1188) to produce bam files. (Li & Durbin, 2009). The bam files are processed using samtools (version 1.21) to produce mapped sorted bam files. (Danecek et al., 2021). The processed bam files are subjected to freebayes (version 1.3.8) and bcftools (version 1.7) to produce normalised variants which are later annotated using snpEff (version 5.2e) with a custom config file (GitHub - Kanyerezi30/EVD_2025). (Cingolani et al., 2012; Danecek et al., 2021; Garrison & Marth, 2012). The annotated normalised variants are subjected to bcftools to produce a consensus genome. Consensus genomes from both ONT and illumina were inspected to produce one final consensus genome.
Results
Sequence output:
Sequencing using the ONT GridION platform generated a preliminary consensus genome with 82% coverage within 2 hours of runtime. After 3 hours of runtime, the coverage reached 85%, which was used for analysis and reporting within 8 hours of the outbreak declaration. After 24 hours, a 91% genome coverage was generated for more comprehensive genomic analyses. Subsequent sequencing using the Illumina MiSeq platform after 48 hours yielded a consensus genome with 90% coverage. Consensus sequences for ONT and Illumina were combined to generate a final consensus at 97% genome coverage for sharing to the pathoplexus (pathoplexus.org) with accession number PP_0011CQ4.1. (Vecchia, 2024).
Data Analysis:
Phylogenetic analysis: To identify the virus species and examine the relationship with historical outbreaks, a phylogenetic analysis was performed which confirmed SUDV for the index case, CL292200/UGA/2025. Phylogenetic reconstruction revealed that the viral genome exhibits a high degree of nucleotide identity and closely clusters with sequences from the 2012 Luwero outbreak in Uganda. Notably, the 2025 strain does not cluster with SUDV sequences from the most recent 2022 outbreak, indicating no direct evolutionary link between the two outbreaks (Figure 2).
These findings suggest that the current outbreak is not a continuation of the 2022 transmission chain and does not support the hypothesis of persistent human-to-human transmission from convalescent individuals or undetected subclinical cases. Instead, the strong phylogenetic relationship with the 2012 Luwero lineage raises the possibility of a shared epidemiological origin, potentially involving a common zoonotic reservoir or an independent spillover event, rather than a direct viral lineage continuation from previously known outbreaks. These data emphasize the need for expanded genomic surveillance to better understand the mechanisms driving SUDV emergence and spillover in Uganda.
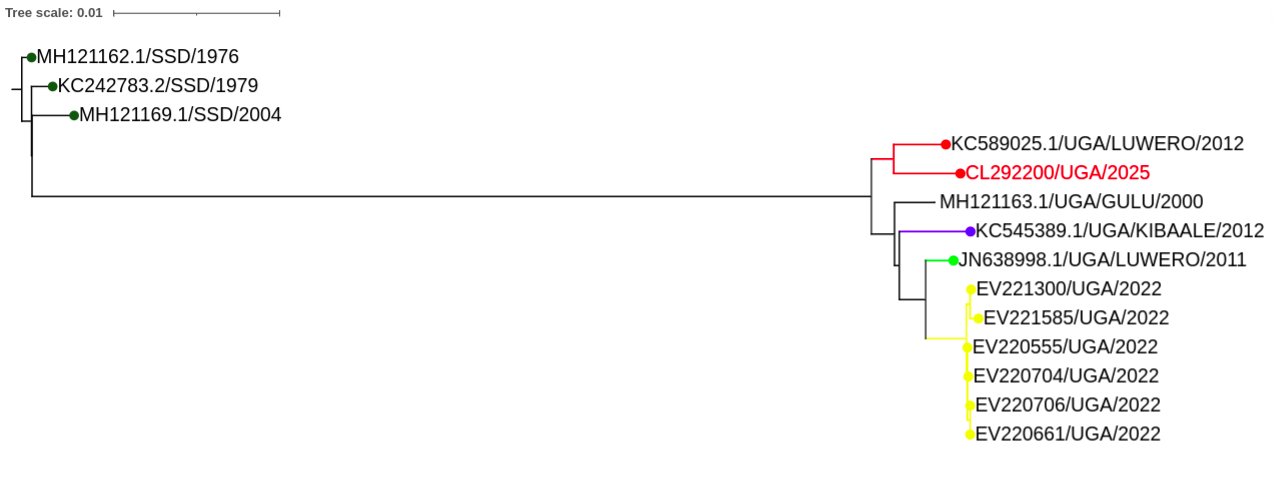
Fig 2: Phylogenetic tree showing the evolutionary relationship between the current (2025) outbreak and past outbreaks. The viral genome of the prevailing outbreak exhibits a high degree of nucleotide identity and closely clusters with sequences from the 2012 Luwero outbreak in Uganda.
Discussion
The present investigation aimed to rapidly characterize the genome sequence of the Ebola virus from the index case in the current outbreak to gain insights into the evolutionary origin of the virus, thereby guiding effective and targeted public health responses. For the first time during an Ebola outbreak in Uganda, we rapidly sequenced, analyzed, and reported the genomic results within 24 hours of the index case identification to support outbreak declaration and an appropriate public health response. This is in alignment with the proposed 7-1-7 global strategy focusing on rapid outbreak detection (within 7 days of emergence), reporting (to public health authorities within 1 day), and control (effective response is mounted within 7 days) (Frieden et al., 2021). Phylogenetic analysis confirmed the SUDV as the etiological agent of the index case (CL292200) in the current outbreak with a high nucleotide identity and close phylogenetic clustering with SUDV sequences from the 2012 Luwero outbreak and no relationship to the 2022 Mubende outbreak, suggesting a shared epidemiological identity with the 2012 event. These findings refute the possibility of sustained human-to-human transmission from the 2022 outbreak and raise questions regarding the current outbreak’s source and transmission dynamics.
The phylogenetic linkage with the 2012 Luwero outbreak implicates a possible independent spillover event and or shared zoonotic reservoir, underpinning the need for intensified ecological surveillance and etiological investigations targeting SUDV’s zoonotic reservoir(s). Understanding spillover mechanisms is crucial for building modern predictive models that might support reservoir identification and monitoring as well as environmental conditions that promote viral disease spread. (Plowright et al., 2017). Furthermore, there is a need to investigate the replication efficiency and transmission potential of SUDV, in addition to any other factors that might affect viral fitness over time, while closely monitoring the contacts of the index case as well as any new confirmed cases. Interestingly, the genetic divergence of SUDV into distinct South Sudan and Ugandan strains is a rare phenomenon among filoviruses and may provide insights into the virus’s interaction with its hosts and environment.
These genomic insights are foundational for guiding epidemiological and ecological investigations. Integrating genomic data into public health strategies is paramount for understanding viral transmission. This highlights the critical need for robust genomic surveillance and rapid genetic analysis to effectively manage outbreaks.
Conclusion
This investigation provides the first genomic evidence from the ongoing 2025 Ebola outbreak in Uganda. Phylogenetic analysis confirms a close relationship with the 2012 Luwero lineage and emphasizes the need for continued sequencing efforts, surveillance, and rapid public health response. This rapid turnaround of genomic information provided sufficient data to inform the immediate response efforts of the Incident Management Teams
Partners and Collaborators
| Name | Institution |
|---|---|
| Dr Susan Nabadda | National Health Laboratory & Diagnostic Services |
| Dr Isaac Sewanyana | Central Public Health Laboratories |
| Dr Henry Kyobe Bosa | Ministry of Health |
| Dr Misaki Wayengera | Ministry of Health |
| Dr. Sofonias Kifle Tessema | Africa CDC |
| Dr Deo Ssemwanga | Uganda Virus Research Institute |
| Mr Peter Van Heusden | South African National Bioinformatics Institute |
| Prof Alan Christoffels | South African National Bioinformatics Institute |
| Dr Gerald Mboowa | Broad Institute of MIT and Harvard |
| Dr Daniel J. Park | Broad Institute of MIT and Harvard |
| Dr Harris Onywera | Africa CDC |
| Dr Collins Kipngetich Tanui | Africa CDC |
| Dr Sarah Wambui Mwangi | Africa CDC |
| Dr Stephen Balinandi | Uganda Virus Research Institute |
| Mr Tebba Andrew | Central Public Health Laboratories |
| Mr Julius Sseruyange | Central Public Health Laboratories |
| Ms Hellen Rosette Oundo | Central Public Health Laboratories |
| Mr Wilson Tenywa | Central Public Health Laboratories |
| Mr Godwin Tusabe Wenka | Central Public Health Laboratories |
| Moses Murungi | Central Public Health Laboratories |
| Ms Jupiter Marina Kabahita | Central Public Health Laboratories |
| Ms Valeria Nakintu | Central Emergency Response Laboratory |
| Ms Caroline Makoha | Central Public Health Laboratories |
| Dr Aloysious Ssemaganda | Central Public Health Laboratories |
| Mr Stephen Kanyerezi | Central Public Health Laboratories |
| Mr Alisen Ayitewala | Central Public Health Laboratories |
References