Joana Isidro1, Vítor Borges1, Miguel Pinto1, Rita Ferreira1, Daniel Sobral1, Alexandra Nunes1, João Dourado Santos1, Verónica Mixão1, Daniela Santos2, Silvia Duarte2, Luís Vieira2, Maria José Borrego3, Sofia Núncio4, Ana Pelerito4, Rita Cordeiro4, João Paulo Gomes1,*.
1 Genomics and Bioinformatics Unit, Department of Infectious Diseases, National Institute of Health Doutor Ricardo Jorge (INSA), Lisbon, Portugal
2 Technology and Innovation Unit, Department of Human Genetics, National Institute of Health Doutor Ricardo Jorge (INSA), Lisbon, Portugal
3 National Reference Laboratory of Sexually Transmitted Infections, Department of Infectious Diseases, National Institute of Health Doutor Ricardo Jorge (INSA), Lisbon, Portugal
4 Emergency and Biopreparedness Unit, Department of Infectious Diseases, National Institute of Health Doutor Ricardo Jorge (INSA), Lisbon, Portugal
contact: [email protected]
Following the (https://pando.tools/t/first-draft-genome-sequence-of-monkeypox-virus-associated-with-the-suspected-multi-country-outbreak-may-2022-confirmed-case-in-portugal/799), we now release 9 additional genome sequences of Monkeypox virus causing a multi-country outbreak. These sequences were obtained from clinical specimens collected from 9 patients on May 15th and 17th, 2022 through high throughput shotgun metagenomics using Illumina technology (see details bellow), with depth of coverage throughout Monkeypox genome ranging from 38x to 508x (mean of 201x).
The rapid integration of the newly sequenced genomes into the Monkeypox genetic diversity, also including the sequence released by USA* (Gigante et al, Monkeypox virus isolate MPXV_USA_2022_MA001, complete genome - Nucleotide - NCBI), allow us to raise the following main observations:
-
The multi-country outbreak most likely has a single origin, with all sequenced viruses released so far* tightly clustering together (Figure 1).
-
Confirmation of the phylogenetic placement unveiled by the first draft sequence Isidro et al,: the outbreak virus belongs to the West African clade and is most closely related to viruses (based on available genome data) associated with the exportation of monkeypox virus from Nigeria to several countries in 2018 and 2019, namely the United Kingdom, Israel and Singapore (1, 2).
-
Still, the outbreak virus diverges a mean of 50 SNPs from those 2018-2019 viruses (46 SNPs from the closest reference MPXV_UK_P2, MT903344.1) (Table 1_2022-05-23.zip (15.0 KB)), which is far more than one would expect considering the estimated substitution rate for Orthopoxviruses (3).
-
As also mentioned by Rambaut (https://pando.tools/t/discussion-of-on-going-mpxv-genome-sequencing/802), one cannot discard the hypothesis that the divergent branch results from an evolutionary jump (leading to a hypermutated virus) caused by APOBEC3 editing (4)
-
We have already detected the first signs of microevolution within the outbreak cluster, namely the emergence of 7 SNPs (Table 2_2022-05-23.zip (10.9 KB)), leading to 3 descendant branches (Figure 1) including a further sub-cluster (supported by 2 SNPs) involving 2 sequences (PT0005 and PT0008). Notably, these two sequences also share a 913bp frameshift deletion in MPXV-UK_P2-010 gene coding for an Ankyrin/Host Range (Bang-D8L); D7L protein (MT903344.1 annotation). Gene loss events were already observed in the context endemic Monkeypox circulation in Central Africa, being hypothesized to correlate with human-to-human transmission (5).
-
This microevolution scenario also suggests that genome sequencing might provide enough resolution to track the virus dissemination in the context of the current outbreak (which could seem implausible for a dsDNA virus).|
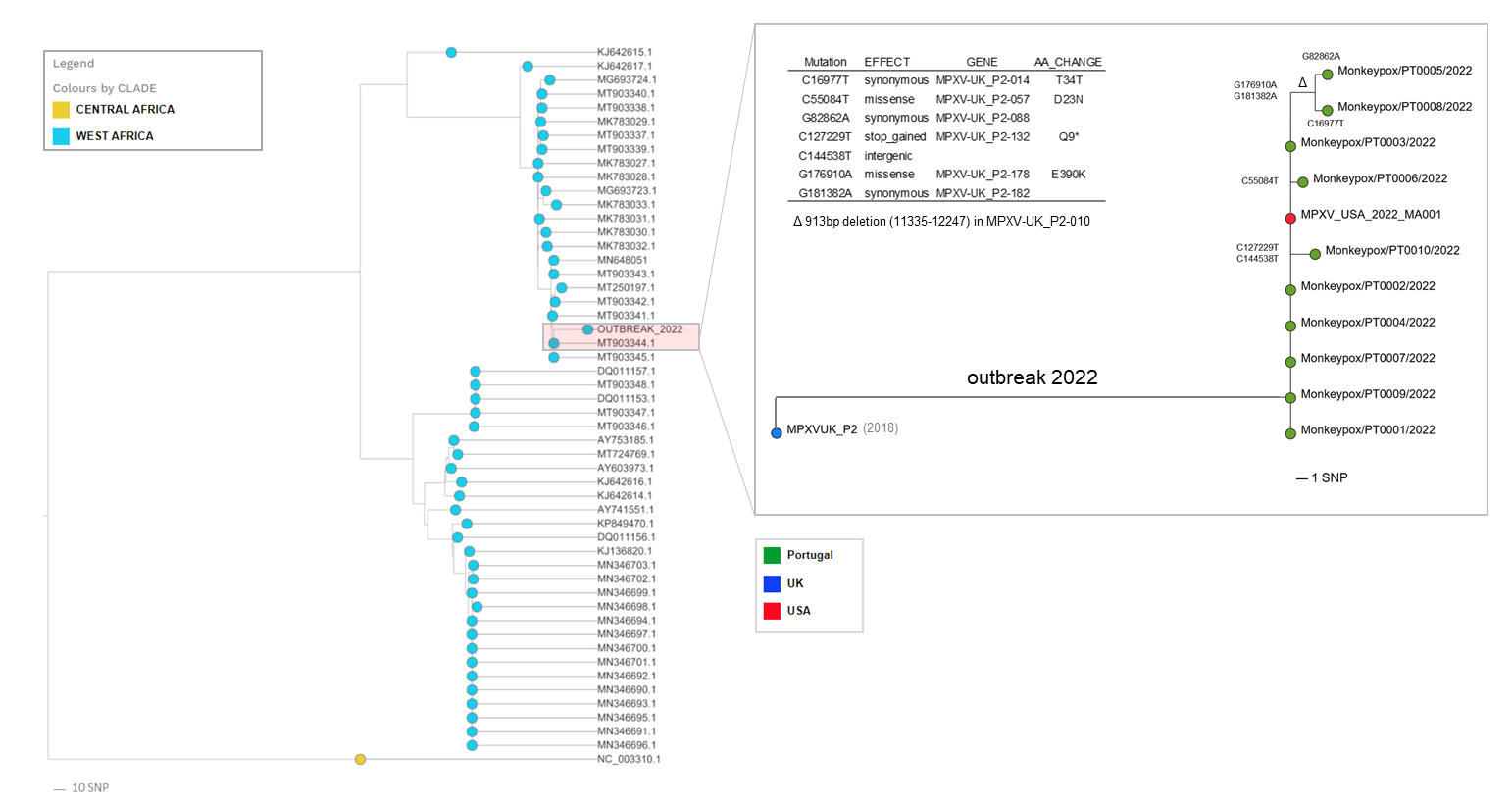
Figure 1. Draft phylogenetic analysis of Monkeypox viral sequences, highlighting the diversity within the outbreak cluster.
‘*’ An additional genome ITM_MPX_1_Belgium was shared by Selhorst et al (ITM) (https://pando.tools/t/belgian-case-of-monkeypox-virus-linked-to-outbreak-in-portugal/801) , but is not currently included in the tree as the sequence most likely has technical issue that might render phylogenetic misplacement.
All sequences from Portugal can be downloaded here
2022-05-23_Monkeypox_PT_sequences.zip (505.7 KB). This dataset includes a further curated version (v2) of the first sequence (Monkeypox/PT0001/2022) and 9 new sequences (Monkeypox/PT0002/2022- Monkeypox/PT0010/2022).
Brief description of the methods
In order to deplete host DNA, skin exudate samples were subjected to sonication and DNAse/RNAse cocktail treatment, prior to DNA extraction with the QIAamp DNA Mini kit (protocol for tissues) (Qiagen). After Nextera XT library preparation, shotgun metagenomics was performed by paired-end sequencing (2x150 bp) on an Illumina NextSeq 2000 apparatus, with about ~40M total reads per sample. Five out of the nine samples were also subjected to ONT MinION sequencing, with generated data being also used to curate the sequences.
Reads were human-depleted and subsequently mapped to the reference MPXV_UK_P2 (MT903344.1; also being used as reference in the newest Monkeypox nextstrain build auspice) using the INSaFLU platform (https://insaflu.insa.pt/). Genome sequences were further curated based on de novo assemblies and careful inspection of mutations. Submission of reads to ENA is ongoing.
- Mauldin MR, McCollum AM, Nakazawa YJ, et al. Exportation of Monkeypox Virus From the African Continent. J Infect Dis. 2022; 225(8):1367-1376. doi: 10.1093/infdis/jiaa559.
- Yinka-Ogunleye A, Aruna O, Dalhat M, et al. Outbreak of human monkeypox in Nigeria in 2017–18: a clinical and epidemiological report. Lancet Infect Dis 2019; 19:872–9. doi: 10.1016/S1473-3099(19)30294-4.
- Firth C, Kitchen A, Shapiro B, et al. Using time-structured data to estimate evolutionary rates of double-stranded DNA viruses. Mol Biol Evol. 2010; 27(9):2038-51. doi: 10.1093/molbev/msq088.
- Pecori R, Di Giorgio S, Paulo Lorenzo J, Nina Papavasiliou F. Functions and consequences of AID/APOBEC-mediated DNA and RNA deamination. Nat Rev Genet. 2022; 7:1–14. doi: 10.1038/s41576-022-00459-8.
- Kugelman JR, Johnston SC, Mulembakani PM, et al. Genomic variability of monkeypox virus among humans, Democratic Republic of the Congo. Emerg Infect Dis. 2014; 20(2):232-9. doi: 10.3201/eid2002.130118.