Vítor Borges1, Joana Isidro1, Mário Cunha2, Daniela Cochicho2, Luis Martins2, Luis Banha2, Margarida Figueiredo2, Leonor Rebelo2, Maria Céu Trindade3, Sílvia Duarte4, Luís Vieira4, Maria João Alves5, Inês Costa6, Raquel Guiomar6, Madalena Santos7, Rita Cortê-Real7, André Dias8, Diana Póvoas8, João Cabo8, Carlos Figueiredo8, Maria José Manata8, Fernando Maltez8, Maria Gomes da Silva9, João Paulo Gomes1,*
Affiliations
1Bioinformatics Unit, Department of Infectious Diseases, National Institute of Health Dr. Ricardo Jorge (INSA), Lisbon, Portugal
2Clinical Pathology – Virology Lab., Instituto Português de Oncologia de Lisboa, Portugal
3Serviço de Hematologia, Instituto Português de Oncologia de Lisboa Francisco Gentil, Lisboa, Portugal
4Innovation and Technology Unit, Department of Human Genetics, National Institute of Health Dr. Ricardo Jorge (INSA), Lisbon, Portugal
5Centre for Vectors and Infectious Diseases Research, Department of Infectious Diseases, National Institute of Health Dr. Ricardo Jorge (INSA), Águas de Moura, Portugal
6National Reference Laboratory for Influenza and other Respiratory Viruses, Department of Infectious Diseases, National Institute of Health Doutor Ricardo Jorge (INSA), Lisbon, Portugal
7Laboratório de Biologia Molecular- Serviço de Patologia Clínica do CHULC, Lisboa, Portugal
8Serviço de Doenças Infecciosas do Hospital de Curry Cabral - CHULC
9Serviço de Hematologia, Instituto Português de Oncologia de Lisboa Francisco Gentil, Lisboa, Portugal
*Corresponding author:
João Paulo Gomes
Department of Infectious Diseases,
National Institute of Health Doutor Ricardo Jorge, Av. Padre Cruz, 1649-016 Lisbon, Portugal. Tel.: (+351) 217 519 241; fax: (+351) 217 526 400.
E-mail: [email protected]
Abstract
Recent studies have shown that persistent SARS-CoV-2 infections in immunocompromised patients can trigger the accumulation of an unusual high number of mutations with potential relevance at both biological and epidemiological levels. Here, we report a case of an immunocompromised patient with a persistent SARS-CoV-2 infection over at least 196 days. Among the 15 SNPs (11 leading to amino acid alterations) and three deletions accumulated during 164 days of infection, four amino acid changes (V3G, S50L, N87S and A222V) and two deletions (18-30del and 141-144del) occurred in the virus Spike protein. Although no convalescent plasma therapy has been administered, some of the detected mutations have been independently reported in other chronically-infected individuals, which sustains a scenario of convergent adaptive evolution. Altogether, these findings show that it is of utmost relevance to monitor the SARS-CoV-2 evolution in immunocompromised individuals, not only to identify novel potentially adaptive mutations, but also to mitigate the risk of introducing “hyper-evolved” variants in the community.
Keywords: SARS-CoV-2, COVID-19, long-term infection, virus evolution, immunocompromised host, genome sequencing
Introduction
Long-term persistence of SARS-CoV-2 in immunocompromised patients has been reported [1-4]. These infections are usually characterized by intermittently detectable SARS-CoV-2 RNA for several months and a within-patient virus evolutionary trajectory marked by accumulation of an unusual high number of mutations [1-4]. Immunocompromised patients are sometimes treated with convalescent plasma and with the anti-viral drug remdesivir, which can trigger viral population shifts, shaping the dynamics of SARS-CoV-2 evolution [1-4]. The first report of persistence and evolution of SARS-CoV-2 in an immunocompromised patient, by Choi and colleagues [1], not only unveiled a remarkable scenario of accelerated viral evolution, but also showed that infectious virus can be recovered from nasopharyngeal samples for several months. In another study, Kemp et al [2] reported a fatal SARS-CoV-2 escape from neutralising antibodies in an immune suppressed patient treated with convalescent plasma. SARS-CoV-2 immune evasion could be linked to the emergence of a dominant viral strain bearing mutations in the key antigen (Spike protein) potentially altering the recognition by antibodies, and thus the sensitivity to convalescent plasma [2]. These and other studies monitoring persistent infections in immunocompromised patients [1-4] have also highlighted the emergence of identical mutations in independent patients, sustaining that persistent RNA positivity can drive convergent adaptive evolution of SARS-CoV-2. Corroborating the potential biological relevance of those recurrent mutations, some of them (e.g., Y453F) are predicted to affect SARS-CoV-2 affinity to ACE-2 receptors or to be potential involved in immune evasion (e.g., ΔH69/ΔV70 and E484K) [2,5-7]. Of note, the emergent and highly concerning SARS-CoV-2 lineage B.1.1.7 (VOC 202012/01) [8], likely originated in the UK, was hypothesized to be a result of virus evolution in a chronically-infected individual (index patient), as revealed by its unusual high genetic divergence (with key amino acid changes predominantly affecting the spike protein) [8].
Here, we report a case of an immunocompromised patient (non-Hodgkin lymphoma patient under immunosuppressive therapy) with SARS-CoV-2 positivity spanning over at least 196 days. Although no convalescent plasma therapy has been administered, the long-term within-patient evolution of SARS-CoV-2 was still marked by a mutation accumulation signature that not only resembles other similar reports, but also highlights novel potentially biologically relevant mutations.
Results
Clinical case
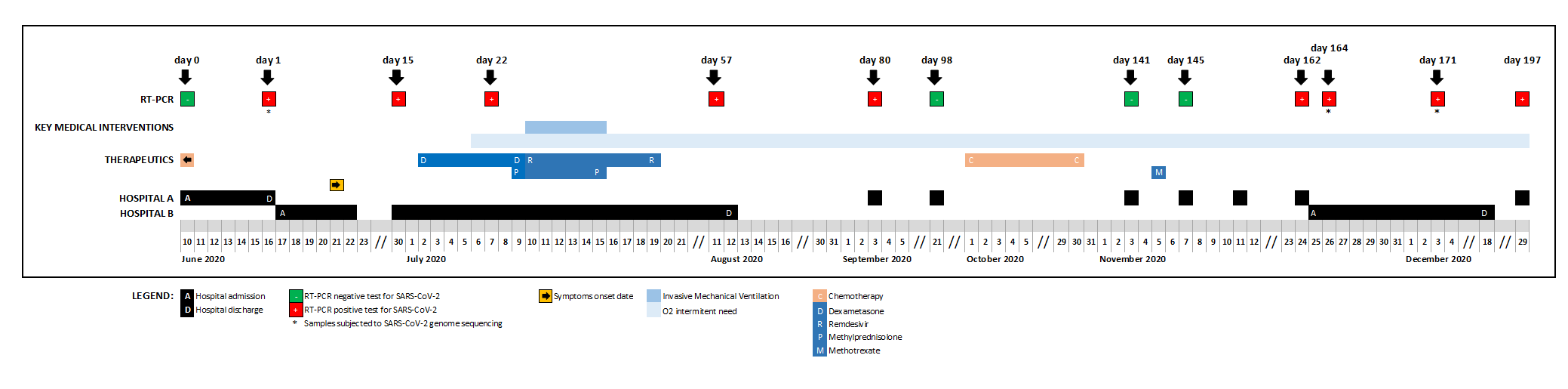
A female patient, aged 61, diagnosed with stage IVB non-Hodgkin diffuse large B-cell lymphoma, was admitted to hospital A on June 10, 2020, due to a bacterial infection in the setting of post-chemotherapy neutropenia, testing negative for SARS-CoV-2 at the admission (Supplementary Figure 1). After the detection of a cluster of SARS-CoV-2 positive cases in that hospital, the patient was again screened for SARS-CoV-2 six days later, testing positive, while being asymptomatic. She was transferred to a COVID-19 ward of Hospital B, where she stayed for 57 days. During this period, she evolved from mildly symptomatic to respiratory insufficiency requiring invasive mechanical ventilation, from which she recovered slowly. She was treated with remdesivir for 10 days and high dose corticosteroids for 7 days during the hospitalization period, being discharged on day 58. During the next months, she remained intermittently symptomatic, ranging from fatigue, cough and low grade fever to shortness of breath requiring supplemental oxygen at home. Residual pneumonitis with extensive fibrotic changes remained evident on computed tomography scan. Meningeal lymphoma progression required weekly intrathecal chemotherapy in October and systemic methotrexate administration on day 143, after which systemic symptoms, cough and dyspnea became more evident. Aiming at further investigation of these symptoms, the patient was readmitted on day 163 in a COVID ward. Bronchoalveolar lavage and transbronchial biopsy were performed and other possible causes were excluded (other respiratory viruses; bacterial causes of atypical pneumonia; Pneumocystis jirovecii pneumonia; non-infectious causes of intersticial pneumonitis / fibrosis). No empirical antibiotic treatment was administered. Exertional dyspnoea requiring supplemental oxygen remained the most prominent clinical feature at discharge. After discharge, intermittent symptoms and partial respiratory insufficiency persisted. Reappearance of neurological symptoms lead again to intrathecal chemotherapy, with partial relief. SARS-CoV-2 RT-PCR positivity was intermittent during the 197-days long-term infection (details in Supplementary Table 1 and Supplementary Figure 1). The patient was negative for antibodies anti-SARS-CoV-2, IgG/IgM (day 171).
Genomic investigation
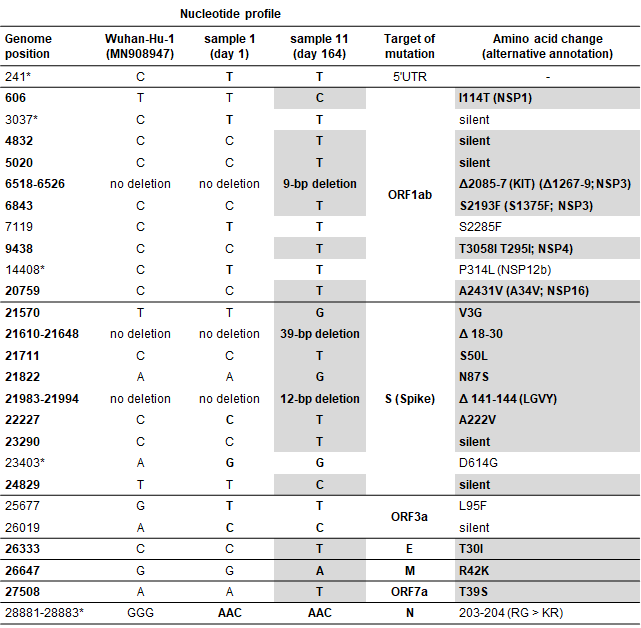
In order to confirm the long-term COVID-19 infection (and exclude the re-infection hypothesis) and monitor SARS-CoV-2 within-patient evolution, viral genome sequencing was performed, as previously described [9], on a nasopharyngeal swab obtained on day 1 and on sputum and bronchoalveolar lavage specimens collected on days 164 and 171, respectively (Supplementary Tables 1 and 2). Although viral culture using the clinical specimen collected on day 164 was also attempted, no virus recovery was achieved. Integration of the viral genome sequence obtained on day 1 (Portugal/PT1525a/2020; GISAID accession EPI_ISL_941339) in the phylogenetic diversity of SARS-CoV-2 in Portugal (SARS-CoV-2 Portugal) confirmed that the immunocompromised patient mostly likely acquired the infection in the context of the nosocomial outbreak detected in Hospital A by mid-late June. In fact, the genome sequence was found to be identical to that of other outbreak-associated inpatients in the same hospital, falling within a cluster also enrolling COVID-19 cases detected at community level (SARS-CoV-2 Portugal) (Figure 1). The outbreak-causing SARS-CoV-2 belongs to COG-UK lineage B.1.1.119 and Nextstrain clade 20B, carrying the Spike amino acid change D614G (Table 1). It diverges from the Wuhan-Hu-1/2019 reference genome (MN908947.3) [10] and the clade 20B root by ten and three SNPs, respectively (Table 1). Analysis of the SARS-CoV-2 genome sequence collected at day 164 (Portugal/PT1525b/2020; GISAID accession EPI_ISL_941340) confirmed the exact ancestral genomic backbone of the virus collected at day 1, with the remarkable addition of three deletions and 15 SNPs (Table 1). Of note, two SNPs (T21570G and C21771T, leading to Spike amino acid changes V3G and S50L) detected in this sample displayed intra-host intermediate frequency (52% and 93%), sustaining that they might be recent emerging mutations. A partial genome sequence could be obtained on day 171, revealing no additional mutations. Among the extra 15 SNPs detected in the evolved SARS-CoV-2, 11 lead to amino acid alterations and the remaining four are silent. In total, four amino acid changes (V3G, S50L, N87S and A222V) and two deletions (18-30del and 141-144del) occurred in the virus Spike protein. Remarkably, as detailed in Table 1, several mutations detected during the long-term infection monitored in this study have been independently observed in similar studies focusing SARS-CoV-2 evolution in chronically-infected individuals.
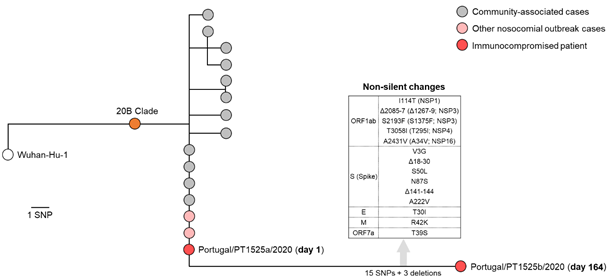
Figure 1. Integration of the viral genome sequences recovered during the long-term SARS-CoV-2 infection of an immunocompromised individual in the phylogenetic diversity of SARS-CoV-2 in Portugal (SARS-CoV-2 Portugal). The immunocompromised patient mostly likely acquired the infection in the context of the nosocomial outbreak detected in Hospital A by mid-late June, as revealed by the detection in day 1 of the same genetic profile observed in other outbreak-associated inpatients of the same hospital. During 164 days of infection, SARS-CoV-2 accumulated 15 SNPs and three deletions (Table 1), including several non-silent mutations in the Spike coding gene.
Table 1. List of mutations accumulated during the long-term SARS-CoV-2 infection of an immunocompromised patient.
Footnotes
Genome positions refer to the reference SARS-CoV-2 Wuhan-Hu-1/2019 sequence (GenBank accession MN908947). Nextstrain 20B clade markers are labeled with an asterisk. Nucleotide/amino acids in bold with white background reflect mutations comparing with Wuhan-Hu-1 reference sequence. Nucleotide/amino acids in bold with gray background indicate mutations accumulated during the long-term infection.
Discussion
In the present study, we report the long-term evolution of SARS-CoV-2 in an immunocompromised patient with non-Hodgkin lymphoma. In line with previous findings [1-4], SARS-CoV-2 underwent accelerated and potentially adaptive evolution within the host, with 18 changes being accumulated after 164 days. This corresponds to a rate of 1.3x10-3 mutations per site per year (i.e., ~40 mutations per genome per year), which exceeds the estimated average rate of evolution of SARS-CoV-2 (around 8x10-4 mutations per site per year, i.e., around 23 mutations per genome per year) [11]. In particular, SARS-CoV-2 evolved some of the exact same mutations seen in other immunocompromised individuals, most of them altering Spike, the key host-interacting protein and antigen. The first remarkable example is the Spike 141-144 deletion (amino acids LGVY). Indeed, this deletion also emerged during SARS-CoV-2 evolution in another immunocompromised patient with non-Hodgkin lymphoma (detection after 132 days) [4], in a patient with severe antiphospholipid syndrome (detection at day 152) [1] and in an asymptomatic immunocompromised patient with cancer (detection after 70 days) [3]. Of note, this recurrent observation of Spike 141-144 deletion in chronically-infected individuals corroborates the highly plausible hypothesis that the emergent SARS-CoV-2 lineage B.1.1.7, which harbors the spike Y144 deletion, resulted of virus evolution in a chronically-infected individual [5]. Another relevant recurrent mutation detected in the present study is the Spike S50L amino acid change, as it was also detected during prolonged COVID-19 in another lymphoma patient [4]. Although no experimental data is available about the functional role of S50L (which falls within the Spike N-terminal domain, NTD), recent computational analysis suggest that it might have strong stabilizing effects on SARS-Cov-2 full-length Spike protein [12]. In the present study, we detected another large deletion in Spike, leading to the loss of amino acids 18-30 (LTTRTQLPPAYTN). This region within NTD might be of particular functional interest as another large deletion in the proximal protein region (amino acids 12-18) was found to emerge 142 days after SARS-CoV-2 evolution in an immunocompromised patient [1]. Outside Spike, we highlight the SNP C9438T leading to T295I amino acid change in NSP4 protein, as this exact mutation was detected during persistence and evolution of SARS-CoV-2 in another patient with non-Hodgkin diffuse B-cell lymphoma [4].
Although the Spike A222V mutation (within NTD) has not yet been reported to recurrently emerge during long-term infections, it has been specially focused by the scientific community. In fact, it is shared by a SARS-CoV-2 lineage (B.1.177; Nextstrain clade 20A.EU1), likely emerging in Spain by summer 2020, that markedly increased in frequency worldwide [13]. In addition, another Spike A222V-bearing variant was the cause of one of the first re-infection cases reported worldwide [14]. A222V is also predicted to be located within one of the epitopes recognized by unexposed humans [15]. Altogether, our results consolidate the expectation that A222V might have a still to be disclosed functional impact on Spike host-interacting activities.
Intriguingly, our report describes accelerated SARS-CoV-2 within-patient evolution in the absence of convalescent plasma therapy. While the lack of plasma-induced selective pressure might explain the lack of mutations affecting the epitope-enriched Spike receptor binding domain (RBD), a rapid viral evolution targeting other key Spike regions was still triggered. Considering that this non-Hodgkin lymphoma patient was under treatment with anti CD20 antibodies before infection and was subjected to intrathecal and systemic chemotherapy during the course of the SARS-CoV-2 infection, it is very likely that the patient was particularly immunosuppressed, thus promoting viral adaptation. In fact, our study highlights novel potentially biologically relevant mutations, while consolidate a scenario of SARS-CoV-2 convergent evolution in immunocompromised individuals, which is a hallmark of adaptive evolution. It also reinforces the need to monitor the virus evolution in immunocompromised individuals, not only to identify novel adaptive traits of SARS-CoV-2, but also to mitigate the risk of introducing “hyper-evolved” adapted variants in the community. For example, the evolution towards alteration of epitopes might have an unpredictable impact on vaccines efficacy. Ultimately, this study highlights the need to revisit the approaches for the follow-up of COVID-19 immunocompromised patients, namely the current recommendations for social confinement.
Methods
Clinical specimens and RT-PCR testing. Multiple respiratory clinical specimens were collected from the immunocompromised patient for SARS-CoV-2 RT-PCR testing during the study period (Supplementary Table 1). Hospital A applied the RT-PCR test Seegene Allplex SARS COV 2 assay (until June 30th: v1 - gene E, RdRP and N; after July 1st: v2 - gene E, RdRP/S and N, analytical sensibility of 50 RNA copies/PCR), while Hospital B used for NP/OP the rRT- PCR test cobas® SARS-CoV-2, analytical sensibility of 32 copies/mL (21–73 copies/ml; CI 95%) (gene E) and 25 copies/mL (17–58 copies/ml; CI 95%) (gene ORF1ab), and, for sputum, the Novel Coronavirus(2019-nCoV) RT-PCR Detection Assay (Fosun 2019-nCov qPCR), analytical sensibility of 300 copies/mL (gene E, ORF1ab and N). SARS-CoV-2 positive RNA samples were sent to the National Institute of Health (INSA) Dr. Ricardo Jorge for SARS-CoV-2 whole-genome sequencing and bioinformatics analysis.
SARS-CoV-2 genome sequencing. Genome sequencing was performed at INSA following an amplicon-based whole-genome amplification strategy using tiled, multiplexed primers [16], according to the ARTIC network protocol (Artic Network; nCoV-2019 sequencing protocol) with slight modifications, as previously described [9]. In brief, after cDNA synthesis, whole-genome amplification was performed using two separate pools of tiling primers [pools 1 and 2; primers version V3 (218 primers) was used for all samples: artic-ncov2019/primer_schemes/nCoV-2019 at master · artic-network/artic-ncov2019 · GitHub]. The two pools of multiplexed amplicons were then pooled for each sample, followed by post PCR clean-up and Nextera XT dual-indexed library preparation, according to the manufacturers’ instructions. Sequencing libraries were paired-end sequenced (2x150bp) on an Illumina NextSeq 550 apparatus, as previously described [9]. All bioinformatics analysis (from reads quality control to variant detection/inspection, sequence consensus generation and minor variants analysis) was conducted using the online (and locally installable) INSaFLU platform (https://insaflu.insa.pt/) [17], as previously described [9]. The genome sequence of SARS-CoV-2 Wuhan-Hu-1/2019 virus (GenBank accession MN908947) was used as reference for mapping and SNV annotation [10]. Regions with depth of coverage below 10-fold were automatically masked in INSaFLU pipeline by placing undefined bases “N” in the consensus sequence. Low coverage regions were visually inspected on “.bam” files using Integrative Genomics Viewer (IGV) and the error-prone position 1871 was excluded. Single nucleotide variants (SNV) were assumed in consensus when they displayed more than 50% of intra-patient frequency. Coronapp (http://giorgilab.dyndns.org/coronapp/) [18] was applied to refine the impact of mutations at protein level. Clade and lineage assignments were performed using Nextclade (https://clades.nextstrain.org/) and Phylogenetic Assignment of Named Global Outbreak Lineages (Pangolin) (https://pangolin.cog-uk.io/) [19], respectively.
Cell culture
The SARS-COV-2 virus isolation attempt was performed in a biosafety level 3 laboratory (BSL3) at INSA. A clinical specimen (sputum, collected on day 164) was used for infecting Vero E6 cells, which were maintained in Eagle’s minimum essential medium MEM; Gibco, UK, supplemented with 10% fetal bovine serum, penicillin (0,6µg/mL) and streptomycin (60µg/mL). The sample was diluted in MEM (2x, 4x, 8x) and 100 µl of each dilution was inoculated onto 25cm3 flasks with a 70% monolayer of cells prepared 24 h before and washed with phosphate-buffered saline (PBS). The inoculated cells were incubated for one hour at 37ºC, 5% CO2, to allow virus adsorption. After that, 10 ml of MEM was added to each flask. The cultures were incubated at 37ºC, 5% CO2 and observed daily for CPE. After 3 days, none of dilutions showed cytopathic effect (CPE). Despite the negative result, a blind passage was made and new cells were infected by repeating the first passage method. Again after 3 days, none of dilutions at the new passage showed CPE.
SARS-CoV-2 serology
In vitro qualitative detection of antibodies to SARS-CoV-2 in human serum was performed using Roche Elecsys Anti-SARS-CoV-2 assay. This assay measures total immunoglobulins directed toward a recombinant nucleocapsid protein from SARS-CoV-2, reporting a ratio of specimen electrochemiluminescent signal to calibrator.
Data availability.
SARS-CoV-2 genome sequences generated in this study were uploaded to GISAID database (https://www.gisaid.org/). Accession numbers are provided in Supplementary table 2.
Integration of the sequence data generated in this study on behalf of the SARS-CoV-2 genetic diversity and geotemporal spread in Portugal can be consulted in SARS-CoV-2 Portugal.
Ethical declaration
Verbal and written informed consent were obtained from the patient to allow the use of the clinical and virological data during prolonged infection. The SARS-CoV-2 genome sequencing study was approved by the Ethical Committee (“Comissão de Ética para a Saúde”) of the Portuguese National Institute of Health.
Acknowledgements
This study is partially co-funded by Fundação para a Ciência e Tecnologia and Agência de Investigação Clínica e Inovação Biomédica (234_596874175) on behalf of the Research 4 COVID-19 call. Some infrastructural resources used in this study come from GenomePT project (POCI-01-0145-FEDER-022184), supported by COMPETE 2020 - Operational Programme for Competitiveness and Internationalisation (POCI), Lisboa Portugal Regional Operational Programme (Lisboa2020), Algarve Portugal Regional Operational Programme (CRESC Algarve2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (ERDF), and by Fundação para a Ciência e a Tecnologia (FCT).
References
- Choi B, Choudhary MC, Regan J, Sparks JA, Padera RF, Qiu X, et al. Persistence and evolution of SARS-CoV-2 in an immunocompromised host. New England Journal of Medicine 2020; 383(23), 2291-2293. doi:10.1056/NEJMc2031364
- Kemp SA, Collier DA, Datir RP, Ferreira IATM, Gayed S, Jahun A, et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 2021. doi: 10.1038/s41586-021-03291-y.
- Avanzato VA, Matson MJ, Seifert SN, Pryce R, Williamson BN, Anzick SL, et al. Case study: prolonged infectious SARS-CoV-2 shedding from an asymptomatic immunocompromised individual with cancer. Cell 2020 ; 183(7), 1901-1912. doi:10.1016/j.cell.2020.10.049
- Bazykin GA, Stanevich O, Danilenko D, Fadeev A, Komissarova K, Ivanova A, et al. Emergence of Y453F and Δ69-70HV mutations in a lymphoma patient with long-term COVID-19. Virological.org 2020. Available from: https://pando.tools/t/emergence-of-y453f-and-69-70hv-mutations-in-a-lymphoma-patient-with-long-term-covid-19/580
- Baum A, Fulton BO, Wloga E, Copin R, Pascal KE, Russo V, et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020; 369(6506), 1014-1018. doi:10.1126/science.abd0831
- Welkers MR, Han AX, Reusken CB, Eggink D. Possible host-adaptation of SARS-CoV-2 due to improved ACE2 receptor binding in mink. Virus Evolution 2021; 7(1): veaa094. doi:10.1093/ve/veaa094
- Greaney AJ, Loes AN, Crawford KH, Starr TN, Malone KD, Chu HY, et al. Comprehensive mapping of mutations to the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human serum antibodies. bioRxiv 2021. doi:10.1101/2020.12.31.425021
- Rambaut A, Loman N, Pybus O, Barclay W, Barrett J, Carabelli A, et al. Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. Virological.org 2020. Available from: https://pando.tools/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563
- Borges V, Isidro J, Cortes-Martins H, et al. Massive dissemination of a SARS-CoV-2 Spike Y839 variant in Portugal. Emerg Microbes Infect 2020; 2:1-58. doi:10.1080/22221751.2020.1844552.
- Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature 2020; 580(7803):E7. doi:10.1038/s41586-020-2202-3
- Hadfield J, Megill C, Bell SM, et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 2018; 34(23):4121–4123. doi:10.1093/bioinformatics/bty407
- Teng S, Sobitan A, Rhoades R, Liu D, Tang Q. Systemic effects of missense mutations on SARS-CoV-2 spike glycoprotein stability and receptor-binding affinity. Briefings in bioinformatics 2020; bbaa233. doi:10.1093/bib/bbaa233
- Hodcroft EB, Zuber M, Nadeau S, Crawford KH, Bloom JD, Veesler D, et al. Emergence and spread of a SARS-CoV-2 variant through Europe in the summer of 2020. MedRxiv 2020. doi:10.1101/2020.10.25.20219063v2
- To KKW, Hung IFN, Ip JD, Chu AWH, Chan WM, Tam AR, et al. Coronavirus disease 2019 (COVID-19) re-infection by a phylogenetically distinct severe acute respiratory syndrome coronavirus 2 strain confirmed by whole genome sequencing. Clinical Infectious Diseases 2020; ciaa1275. doi:10.1093/cid/ciaa1275
- Mateus J, Grifoni A, Tarke A, Sidney J, Ramirez SI, Dan JM, et al. (2020). Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science 2020; 370(6512), 89-94. doi: 10.1126/science.abd3871
- Quick J, Grubaugh ND, Pullan ST, et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc 2017; 12(6):1261–1276. doi:10.1038/nprot.2017.066
- Borges V, Pinheiro M, Pechirra P, Guiomar R, Gomes JP. INSaFLU: an automated open web-based bioinformatics suite “from-reads” for influenza whole-genome-sequencing-based surveillance. Genome Med 2018; 10(1):46. Published 2018 Jun 29. doi:10.1186/s13073-018-0555-0
- Mercatelli D, Triboli L, Fornasari E, Ray F, Giorgi FM. Coronapp: A web application to annotate and monitor SARS-CoV-2 mutations. J Med Virol 2020; https://doi.org/10.1002/jmv.26678
- Rambaut A, Holmes EC, O’Toole Á, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol 2020; 5(11):1403-1407. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology | Nature Microbiology
- Hodcroft EB. 2021. “CoVariants: SARS-CoV-2 Mutations and Variants of Interest.” https://covariants.org/
Supplementary Figure 1. Timeline of key clinical events during the long-term SARS-CoV-2 infection of an immunocompromised patient with non-Hodgkin lymphoma.
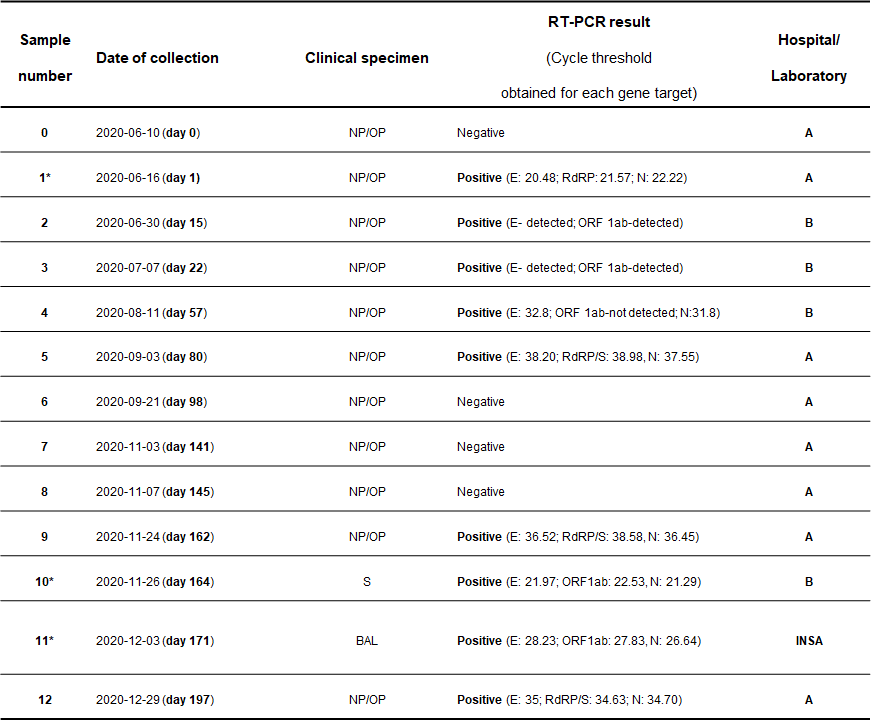
Supplementary Table 1. Description of clinical samples subjected to SARS-CoV-2 RT-PCR test.
Footnotes
- Samples subjected to SARS-CoV-2 genome sequencing; OP: oropharyngeal swab; NP: nasopharyngeal swab; S: sputum; BAL: Bronchoalveolar lavage
Supplementary Table 2. Sequencing data of SARS-CoV-2 genome consensus sequences obtained at day 1 and day 164.

Footnotes
- Regions with depth of coverage below 10-fold were automatically masked in INSaFLU pipeline by placing undefined bases “N” in the consensus sequence. One of these small regions (22986-23122) in the Portugal/PT1525b/2020 sequence falls within S gene (coverage between 2- and 9-fold). Due to its biological relevance, all reads were inspected, showing no differences for the reference genome (this region was then unmasked).