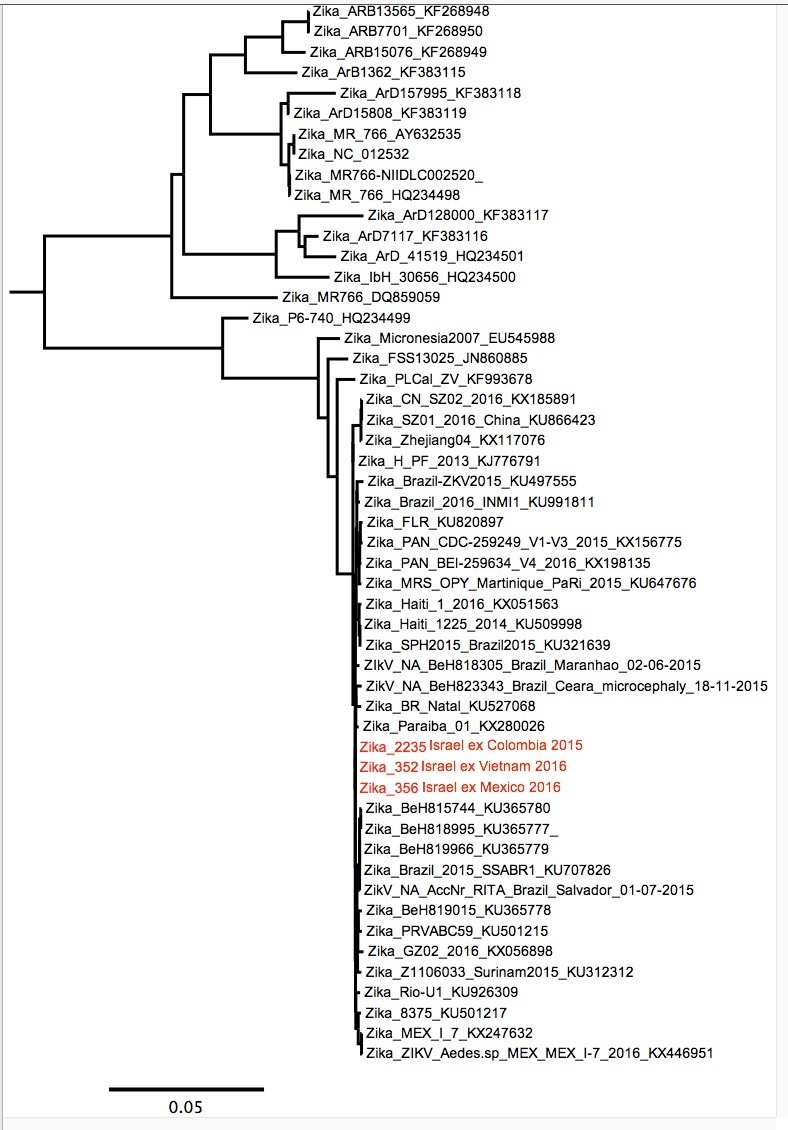
There’s a paper in EID: http://wwwnc.cdc.gov/eid/article/22/8/16-0480-f1 which shows a Vietnamese E gene fragment (pos 1304-1534 in reference) in a returning Israeli traveller which uses BEAST to place the sequence inside the South American part of the tree rather than further down in the SE Asian part where we’d expect it. The authors make nothing of this, even though it could potentially suggest that American Zika has looped back into SE Asia. My summer student Mark has been looking at this again using MEGA and has found that all methods confirm the result - except that we now have the French Polynesian sequence in the wrong place (the published BEAST tree has it in the right place). MP tree below. So, we are forced to conclude that it might just be lack of phylogenetic signal in the 231 bp (even though the max p-dist is 15%, there are 62 variant position of which 12 involve non-synonymous substitutions).

I had an email conversation with the authors on 7-11th July about this point. There isn’t enough phylogenetic signal to determine the placement of the Vietnam strain and thus the direction of movement.They seemed to understand the issue but I think it was too late to make any changes to the manuscript. Contact [email protected] if you want to discuss further.
The sequence of the Israel isolate is just 231 bases and there is essentially no information in that short region of the genome which could differentiate between the South American viruses which recently came out of Asia, and the Asian viruses.
I aligned the new 231-base sequences (the one from the Vietnam traveler
and two in Israel who were infected in South America) to the complete
genomes of representative isolates, but the VIROLOGICAL site does not allow me to upload the file here. Send me an email if you want the alignment.