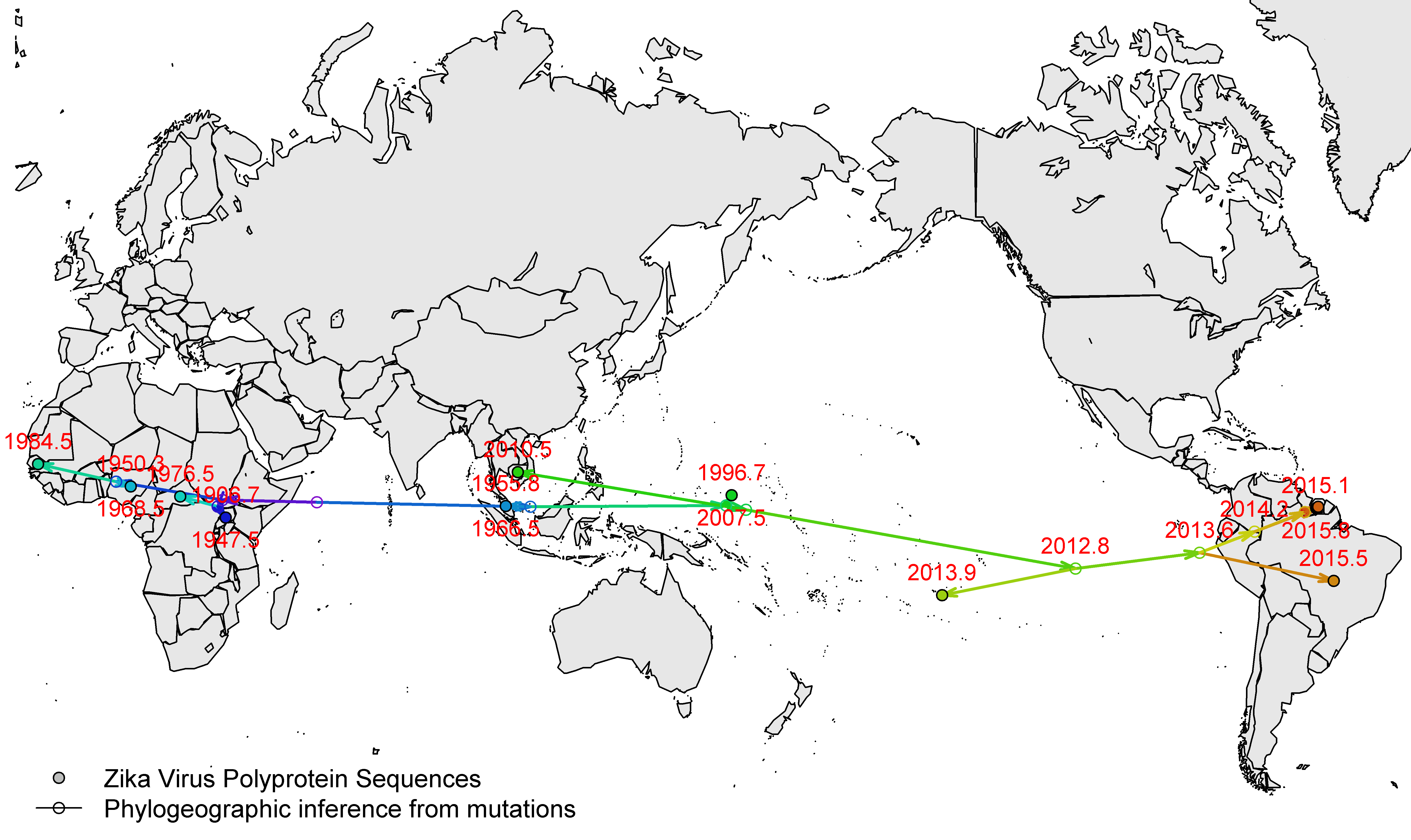
There were a few polyprotein sequences on Genbank (28 Jan 2016), and 12 had good dates, so here is an initial timescaled tree (HKY+Gamma4, relaxLn, constant population).
Sam, I am very concerned about the integrity of some of the Zika sequences available on GenBank, particularly the E gene. Some have a complex passage history and others fall in strange places in the phylogeny or have dubious branch lengths. I believe a number of these genomes are being re-sequenced and some should probably be ignored. So, I would not try to infer too much on these data at present. However, it is very clear that the South American outbreak strains are genetically very close to those to recently circulating in Asia.
I agree with Eddie - some of the (older?) sequences on Genbank are definitely wacky. The 766 strain has been sequenced many times, but is very different across sequencing efforts - some of the variants have been passaged >100 times in TC so that might explain why.
@samantha_lycett I certainly like your two figures better than the trees shown in Figure 1 of the recent Institut Pasteur paper (Lancet 2016) and CDC paper (EID 2016). In both of those cases, I suspect their trees were mid-point rooted (or maybe not even rooted–neither paper describes any rooting methodology) and don’t make as much intuitive sense. I was wondering when @evogytis and @arambaut might reprise their PLoS Curr Outbreaks 2014 album, but it looks like you’ve done it already.
@edward_holmes, @Kristian_Andersen – I’d love to hear more about the errors you suspect from old assemblies. If they’re TC/passage-induced mutations, that’s fine (what can you do), but if you think there are actual assembly errors due to repetitive bits of the genome, etc, it’d be nice to know what to keep an eye out for in the future (certain problematic regions). Did you highlight the E gene because it’s naturally highly polymorphic or because of some other property?
Rooting between the Asian and Australian lineages is the standard plan for Zika and probably fine.
I don’t think there are assembly errors but some of the old strains are heavily passaged and likely mouse adapted. As I said, I know that there is a large sequencing effort going on at the moment. Hence, I would hold off detailed phylogeographic analysis etc. until there has been a bit of a refresh.
I will try and post of some of the figures I have done if I can figure out how to do this.
Here’s a genome tree with the amino acid changes mapped onto some of the more interesting branches (numbered according the polyprotein). I can’t 100% guarantee that I have the gene assignment right. Also, I’ve used one of the Uganda/1947 sequences but you can debate which one is the more accurate.
And here’s an env tree. Clearly some of these sequences fall in very odd positions (spot for yourselves) and the root-to-tip regression is all over the place.
To build on @dpark 's point, I’d also be mindful of sequencing artifacts when using the new Brazilian sequences for analyses. Given the high similarity between different isolates (>99.5%) even a small number of sequencing artifacts could be problematic. For example, KU321639 has an insertion at 10,446 in a 6C homopolymer that is almost certainly an artifact.
I’m not sure if this is true throughout S. America, but my understanding is that Ion Torrent is the most popular platform in Brazil in part because of a solid supply chain and better sales/marketing/support. Anecdotally, I’ve had a lot of problems with my collaborators getting Illumina supplies to Brazil in a timely manner. So I’d expect a lot of the upcoming sequenes from Brazil will be Ion and should be carefully evaluated for artifacts.
It would be cool if PGM people made the raw sequencing reads available so issues like this can be corrected or masked where possible. It’s usually easy to spot the length where uncertainty kicks in from the underlying alignments.
An April 2016 paper by Shu Shen et al hypothesizes that the Asian virus came from Africa much more recently than most other analyses have indicated. Shen2016_ZikaVirus.pdf (1.7 MB)
I have not had a chance to obtain the alignments they used, or analyze the same regions of the genome they used (my trees are based on complete genomes), but they seem to have noted that KF383113 and KF383020 belong to different lineages despite both being labelled as Cote d’Ivore 1980.
I had an undergrad project student who was intending to try to replicate that result, but the problem was that Shen et al use MIGRAPHYLA and it became apparent that reconstructing the MIGRAPHYLA pipeline was not really an out-of-the-box install, and beyond the capacity of an undergrad. I don’t think Figure 4 is plausible. Having Senegal as the source of multiple independent dispersals ranging from 1966 to now, isn’t really plausible.
Looks like some serious outgroup rooting issues (similar to that discussed in http:doi.org//10.1371/currents.outbreaks.84eefe5ce43ec9dc0bf0670f7b8b417d ). With a very divergent outgroup the position that falls in the ingroup is essentially arbitrary and the inferences are entirely dependent on it. On the other hand the sequence data from before the 90s are likely to be lab passaged strains, susceptible to contamination and lab adaptation mutations. I would also be very sceptical of the ‘recombination’ events as they are single viruses (i.e., no evidence the mosaic pattern was transmitted). Likely lab generated artifacts as usual.
Here is a project idea for a student - go through each of the Zika sequences so far and try to work out what problems they each have - recurrent NS mutations suggesting cell adaptation, mosaics on primer boundaries, likely contamination because two supposedly temporally or spatially separated sequences are identical and sequenced in the same lab, etc. Mike Worobey did something similar for supposedly old flu sequences a while back.
I would bet that Andrew’s idea that outgroup rooting is the problem, is the correct answer. I don’t think it is necessary to reproduce the results using MIGRAPHYLA, it would be good to reproduce the trees with and without Spondweni virus as the outgroup. The tree at the top of this thread, showing a pre-1900 common ancestor for the Asian/American and African lineages seems certain to be the correct topology given what is known about Zika in Asia in the WWII era. I built trees from complete genomes of Zika plus Spondweni and Dengue and other viruses and my trees also misrooted the Zika clades when those other viruses were in the alignment. I am not sure if it is a classic “long branches attract” problem, but it might be because there is more diversity in the African clades than within the Asia/Americas clade.