Initial observations about putative APOBEC3 deaminase editing driving short-term evolution of MPXV since 2017.
An updated version of this report is now available as a preprint at:
https://www.biorxiv.org/content/10.1101/2023.01.23.525187v1
This document is an initial report on the observation of an abundance of specific mutations in the 2022 MPXV outbreak and related virus genomes that can be ascribed to the action of APOBEC3 host enzymes. It should be considered work in progress and we plan to add additional analysis and interpretation. We also welcome discussion in the thread below. The analyses here are made possible by the groups and researchers who have shared MPXV genome sequence data (Table 1).
Áine O’Toole & Andrew Rambaut
Institute of Evolutionary Biology
University of Edinburgh
Edinburgh, UK
The first MPXV genome sequences from monkeypox cases in 2022 (Isidro et al. 2022; Selhorst et al. 2022) showed, phylogenetically, that these viruses had descended from a clade sampled in 2017-2019 from cases diagnosed in Singapore, Israel, Nigeria and the UK. Comparing 2022 genomes from Portugal, Belgium, USA, Australia, and Germany (see Table 1) with the closest earlier genomes (denoted UK_P2 and UK_P3), identified 47 shared single nucleotide differences (Figure 1).
The long term evolutionary rate of the related variola virus (VARV; the smallpox virus) has previously been estimated to be about 9x10-6 (with 95% credible intervals of 7.8x10-6 – 10.2x10-6) substitutions per site per year (Firth et al. 2010) translating into about 1-2 nucleotide changes per year for a nearly 200,000 nucleotide genome. This makes 47 substitutions in the space of 3-4 years an unexpectedly large number. As MPXV is considered a zoonotic virus with limited human to human transmission, this long branch may be evidence of adaptation to humans allowing for the sustained transmission that is now observed.
However, 42 out of 47 of these nucleotide changes are of a particular type, a dinucleotide change from TC→TT or its reverse complement GA→AA. This specific mutation is characteristic of the action of the APOBEC3 family of deaminases. These act on single stranded DNA to deaminate cytosine to uracil causing a G→A mutation in the other strand when it is synthesised. Most human APOBEC3 molecules have a strong bias towards deaminating 5’TC dinucleotides , with the exception being APOBEC3G which prefers 5’CC dinucleotides (Yu et al. 2004).
Of the remaining 5 nucleotide changes between 2018 and 2022, 3 are GG→AG compatible with negative strand deamination and in the context preferred by APOBEC3G. A previous study showed that the related vaccinia virus was not significantly restricted by APOBEC3G (Kremer et al. 2006) and the low number of mutations at dimers preferred by this enzyme would seem to confirm this. The final 2 mutations are not the result of deamination of cytosine. Given the estimated rate of evolution for variola viruses between 2 and 5 substitutions would be within the expectation for 4 years of evolution. This could indicate that the 42 G→A and C→T changes were the result of bouts of deamination during virus replication due to host antiviral defences.
Genome sequencing of MPXV, including from archived samples and infections in non-human animals, has identified three distinct phylogenetic clades, although limited sampling means this may not be representative of the diversity present in the non-human animal reservoir host. The 2022 human outbreak is part of a clade that has previously been sampled in Nigeria or in cases with travel history in Nigeria with the oldest genome being from 1971 (accession number: KJ642617; (Nakazawa et al. 2015) with the remainder first sampled since 2017 (Mauldin et al. 2022). Examining the phylogenetic tree of these recent genomes also shows a predominance of the same GA→AA and TC→TT mutations suggesting that APOBEC3 deamination is the primary source of observed single nucleotide genetic variation in this group of MPXVs (Figure 1).

Figure 1 | A phylogenetic tree of MPXV genomes sampled from human infections from 2017-2022, with an outgroup sequence from an outbreak in Nigeria in 1971. SNPs along each branch are indicated with circles coloured by whether a mutation is putatively APOBEC edited
TC→TT(blue) andGA→AA(orange), whether it is a non-targetC→TorG→Amutation (teal), or other SNP type (grey).
This observation prompts a number of questions: Is this putative APOBEC3 editing occurring in a non-human animal reservoir host prior to emergence into humans in limited chains of human transmission? Or does this tree represent a multi-year history of sustained human transmission? Is the action of APOBEC3 acting as a driver of adaptation to humans as a host?
To address the first question we considered the wider context at which the putative deamination of cytosine was occurring (Figure 2) on the branch leading to the 2022 outbreak. Although there is some signal that T is not favoured immediately 3’ of the target cytosine, and G is not favoured to the 5’ of the target, there is not strong evidence of a wider preferential context. Studies have shown that the single APOBEC3 enzyme found in rodents exhibits a preference for a 5’TYC (Conticello et al. 2005), a context only seen for 46 of the 142 putative APOBEC3 substitutions across the entire tree.
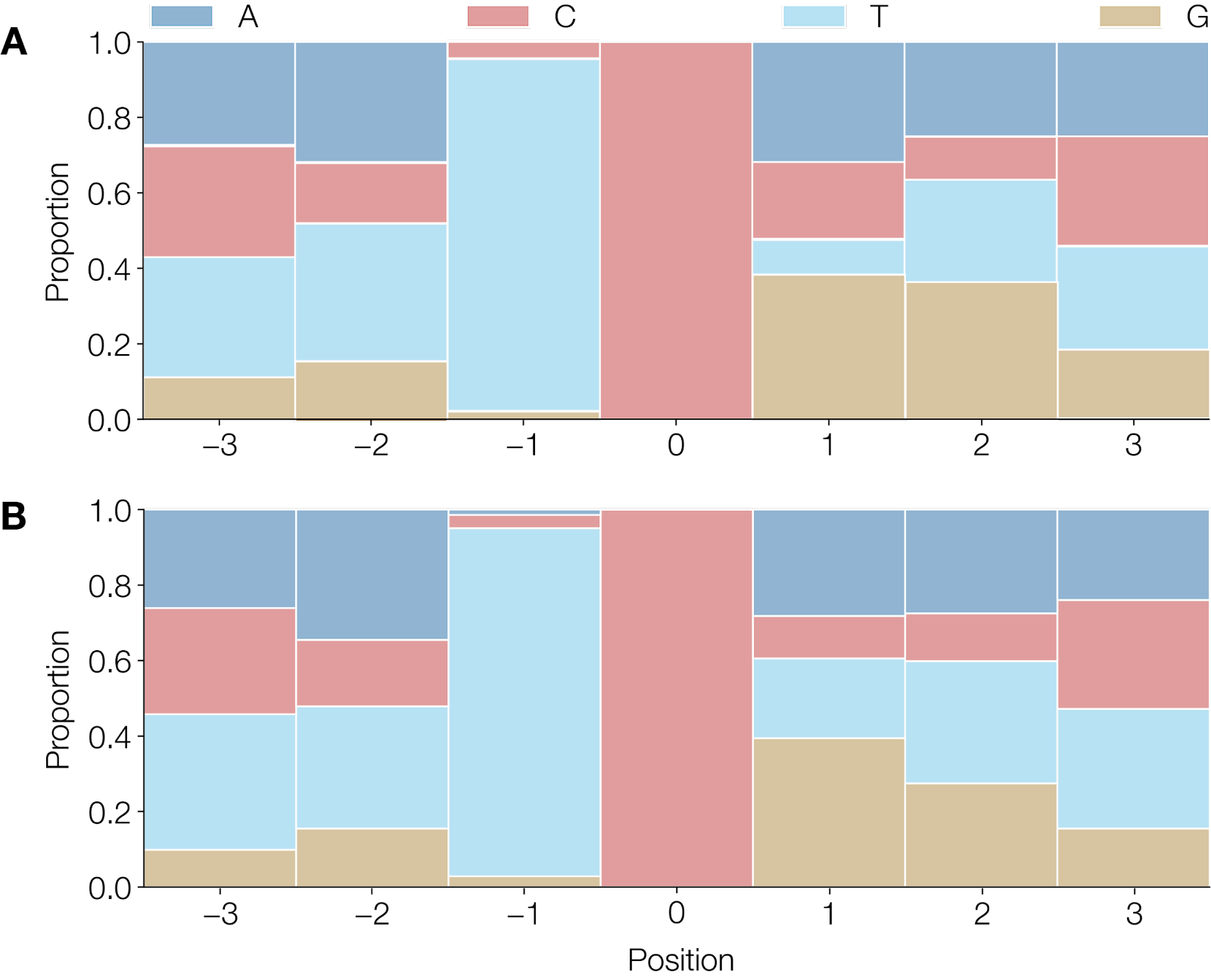
Figure 2 | A) Observed heptamers that show a
C→TorG→Amutated site in 44 of 47 mutations between the UK 2018 sequence and the CDC sequence of the current outbreak (accession numbers MT903345 and ON563414.2 respectively). B) Observed heptamers in 142 of 183 unambiguous mutations that showC→TorG→Amutated sites, normalised for strand across the entire tree excluding the mutations that occur on the 1971 branch. Heptamers associated withG→Amutations have been reverse-complemented to reflect deamination on the negative strand.
If APOBEC3 deamination is characteristic of replication in humans then we would expect to see very little evidence of it prior to the 2017 outbreak as this would primarily represent replication in the non-human reservoir. To examine this, we selected a further outgroup from Liberia, 1970 (accession number DQ011156.1; Likos et al. 2005) and identified 28 mutations that occur on the branch leading to the common ancestor of the 2017 MPXV genomes (Figure 3A). The mutations on this branch do not show such a strong signal, with only 10 of 28 SNPs matching the signature for APOBEC3 editing (Figure 3B). We therefore suggest that the pattern we see in these MPXV genomes since 2017 is indicative of replication in humans and the inheritance of the specific changes that occurred between 2017 and 2018 and then in the viruses from 2022 means that there has been sustained human to human transmission since at least 2017. The 10 mutations that do fit the APOBEC3 profile may represent an additional period of human to human transmission prior to the 2017 cases. Given that this is fewer APOBEC3 type mutations than seen in the branch between 2017 and 2018 (branch C in Figure 1), it is not likely that this represents a long period.

Figure 3 | SNPs that are unique to the branch leading to the common ancestor of the ingroup of the phylogeny in Figure 1 (2017 outbreak of MPXV) . A) Using an MPXV genome from Liberia, 1970 (accession number DQ011156; Likos et al. 2005) and the 1971 MPXV sequence (accession number KJ642617; Nakazawa et al. 2015) as outgroups, we identified mutations that occurred on the branch leading to the common ancestor of the 2017 MPXV genomes. These SNPs fit the mutational pattern described, with the two outgroup sequences sharing the same nucleotide sequence and the 2017 MPXV genomes sharing a distinct variant. B) Only 10 of the 28 mutations on this branch have the mutational signature of APOBEC editing.
Is APOBEC3 driving the fine-scaled evolution of MPXV in humans?
The normal action of APOBEC3 enzymes is anti-viral. Inducing C→T mutations at random locations across the genome is very likely to produce sufficient deleterious changes to inactivate the virus. Retroviruses seem to be a specific target of some APOBEC molecules (hA3G and hA3F) which can be packaged in the virion and then act as the reverse-transcriptase synthesises the first DNA strand from the RNA genome template (Harris et al. 2003; Zhang et al. 2003). The DNA copy is then integrated into the host genome replete with APOBEC induced mutations (G→A in the positive strand) meaning that all resulting progeny RNA genomes will have these mutations and will likely be lethally degraded. It is notable that HIV has a defence against APOBEC3 in a protein, vif, which degrades APOBEC3 and thus the effects are only seen in viruses where vif is defective resulting in ‘hypermutated’ viruses.
With double stranded DNA viruses APOBEC3 enzymes will act as the viral genome is being replicated and single strands are exposed. During repeated rounds of replication either strand can be deaminated leading to both C→T and G→A changes on the positive strand as seen here. Thus it is likely that the genomes that are extensively mutated by APOBEC3 will simply not be viable and will not be transmitted further. The extensive rounds of genome replication that MPXV undertakes in the cytoplasm may mean that most genomes are not affected by APOBEC3 action. Occasionally however a genome that is only modestally mutated by APOBEC3 may remain viable and be transmitted.
Considering all GA and TC dimer sites in the protein coding regions of the 2018 genome (‘MPXV-UK_P3’; accession number MT903345; Mauldin et al. 2022) – i.e., those that could be the target of APOBEC3 editing but had not been by that point – we looked at what the effect of a mutation at these sites would be (Figure 4). Of the 21,230 such dimers, 69.3% (14,707) would produce amino acid replacements, 24.7% (5253) would be synonymous, and 6.0% (1270) would induce stop codons. For the 2022 genomes, of the 40 mutations at these dimers that did occur, 60% (24) were amino acid replacements and 40% (16) were synonymous (a further 2 APOBEC3 mutations were in intergenic regions). The probability of getting 16 or fewer synonymous mutations under a binomial distribution with an expected rate of 0.247 is P=0.024. This supports the hypothesis that what we observe are the residual least harmful APOBEC3 mutations after natural selection has eliminated those with substantial fitness costs to the virus.

Figure 4 | A) Consequence of hypothetical APOBEC3 mutations at target dimer site (either the
Cin theTCtarget site orGin theGAtarget site) in currently unmutated sites in the 2018 MPXV genome (‘MPXV-UK_P3’; accession MT903345; Mauldin et al. 2022) and those observed APOBEC3 mutations on a representative sequence of the current outbreak (specifically, ‘MPXV_USA_2022_MA001’; accession ON563414; Gigante et al. 2022). These are categorised into synonymous (amino acid remaining unchanged), non-synonymous (altered amino acid) or nonsense (editing producing a stop codon).
Does the long branch represent adaptation to humans?
The ‘repertoire’ of mutations that APOBEC3 is able to provide as genetic variation on which natural selection can act is severely restricted. Only a limited number of dinucleotide contexts are present, and the amino acid changes that APOBEC3 editing can induce is also limited (Figure 5). Only 13 different amino acid replacements are possible and they are not reversible by the same mechanism. This means that the chance that a mutation that confers a benefit to the virus is amongst the ones available through APOBEC3 editing is relatively small.

Figure 5 | Amino acid mutations at AA and TT dimer sites in a reference MPXV genome (accession number JX878407) that would have occurred had unobserved APOBEC3 editing occurred to give rise to these. A) Barplot of amino acid changes categorised by Grantham Score (0-50 conservative, 51-100 moderately conservative, 101-150 moderately radical, >150 radical). B) Hypothetical amino acid changes had sites with
AAandTTdimers had they been the result of APOBEC3 editing. Hypothetical mutations originating from stop codons excluded from analysis.
In most studies of APOBEC3 editing (and indeed, other host enzymes that induce mutations through deamination of nucleotides such as ADAR) the induced mutations are clustered and frequent, the presumption being that an APOBEC molecule when present repeatedly deaminates targets. For APOBEC3G in vif-defective HIV-1, it is estimated that only a few APOBEC molecules are packaged in the virion but induce large numbers of mutations (Armitage et al. 2008, 2012). Although there is some clustering of the observed mutations in the 2022 genomes they are generally seen across the entire genome (Figure 6). It is thus likely that this represents multiple episodes of APOBEC3 editing each of which had limited fitness consequences on the virus but conversely these mutations did not occur entirely independently. This non-independence may adversely affect the ability of molecular clock models to estimate time-calibrated phylogenetic trees. The non-reversibility of APOBEC3 induced mutations will also mean that certain phylogenetic substitution models will be more appropriate than others (i.e., non-reversible continuous time Markov chain models or Dollo model may be applicable).

Figure 6 | Distribution of mutational differences between UK_P3 and a representative genome from the 2022 outbreak.
A final consideration is that the constant pressure of APOBEC3 editing, despite MPXV being apparently largely robust to its antiviral effects in humans, may mean that moderately deleterious mutations may accumulate. This may result from genetic drift resulting from the diversity bottleneck at transmission. Given the directional nature of these mutations this may act as a ratchet, irreversibly accumulating fitness-reducing mutations with other mutational processes, occurring at a much lower rate, unable to overcome this. Indeed, additional APOBEC3 type mutations have been observed within 8 genomes sampled from the outbreak in Portugal (Isidro et al. 2022) including 2 that are shared by 2 genomes suggesting ongoing transmission of these.
The analysis presented here suggests further research including sequencing and analysis of within-host variation in APOBEC3 editing to investigate its potential for controlling or moderating MPXV human infections. Variation in APOBEC3 editing within a single individual (perhaps at samples from different lesions) might suggest that it is a significant burden on MPXV.
Table 1 | MPXV genome sequences used in this study.
Accession Country Year Reference KJ642617 Nigeria 1971 (Nakazawa et al. 2015) DQ011156 Liberia 1970 (Likos et al. 2005) MK783031 Nigeria 2017 (Yinka-Ogunleye et al. 2019) MK783029 Nigeria 2017 (Yinka-Ogunleye et al. 2019) MK783027 Nigeria 2017 (Yinka-Ogunleye et al. 2019) MK783028 Nigeria 2017 (Yinka-Ogunleye et al. 2019) MK783030 Nigeria 2017 (Yinka-Ogunleye et al. 2019) MK783033 Nigeria 2017 (Yinka-Ogunleye et al. 2019) MK783032 Nigeria 2017 (Yinka-Ogunleye et al. 2019) MT903339 Nigeria 2017 (Mauldin et al. 2022) MT903340 Nigeria 2017 (Mauldin et al. 2022) MT903338 Nigeria 2017 (Mauldin et al. 2022) MG693724 Nigeria 2017 (Faye et al. 2018) MN648051 Israel 2018 (Cohen-Gihon et al. 2020) MT903342 Singapore 2019 (Mauldin et al. 2022) MT250197 Singapore 2019 (Yong et al. 2020) MT903341 Nigeria 2018 (Mauldin et al. 2022) MT903343 UK 2018 (Mauldin et al. 2022) MT903344 UK 2018 (Mauldin et al. 2022) MT903345 UK 2018 (Mauldin et al. 2022) ON563414.2 USA 2022 (Gigante et al. 2022) UZ-REGA-1 Belgium 2022 (Vanmechelen et al. 2022) ON568298 Germany 2022 (Antwerpen et al. 2022) PT0001 - PT0010 Portugal 2022 (Isidro et al. 2022)
References
Antwerpen, Markus H., Daniel Lang, Sabine Zange, Mathias C. Walter, and Roman Wölfel. 2022. “First German Genome Sequence of Monkeypox Virus Associated to Multi-Country Outbreak in May 2022.” Virological. May 24, 2022. https://pando.tools/t/812
Armitage, Andrew E., Koen Deforche, Chih-Hao Chang, Edmund Wee, Beatrice Kramer, John J. Welch, Jan Gerstoft, et al. 2012. “APOBEC3G-Induced Hypermutation of Human Immunodeficiency Virus Type-1 Is Typically a Discrete ‘All or Nothing’ Phenomenon.” PLoS Genetics 8 (3): e1002550.
Armitage, Andrew E., Aris Katzourakis, Tulio de Oliveira, John J. Welch, Robert Belshaw, Kate N. Bishop, Beatrice Kramer, Andrew J. McMichael, Andrew Rambaut, and Astrid K. N. Iversen. 2008. “Conserved Footprints of APOBEC3G on Hypermutated Human Immunodeficiency Virus Type 1 and Human Endogenous Retrovirus HERV-K(HML2) Sequences.” Journal of Virology 82 (17): 8743–61.
Cohen-Gihon, Inbar, Ofir Israeli, Ohad Shifman, Noam Erez, Sharon Melamed, Nir Paran, Adi Beth-Din, and Anat Zvi. 2020. “Identification and Whole-Genome Sequencing of a Monkeypox Virus Strain Isolated in Israel.” Microbiology Resource Announcements 9 (10). DOI: 10.1128/MRA.01524-19
Conticello, Silvestro G., Cornelia J. F. Thomas, Svend K. Petersen-Mahrt, and Michael S. Neuberger. 2005. “Evolution of the AID/APOBEC Family of Polynucleotide (deoxy)cytidine Deaminases.” Molecular Biology and Evolution 22 (2): 367–77.
Faye, O., C. B. Pratt, M. Faye, G. Fall, J. A. Chitty, M. M. Diagne, M. R. Wiley, et al. 2018. “Genomic Characterisation of Human Monkeypox Virus in Nigeria.” The Lancet Infectious Diseases 18 (3). DOI: 10.1016/S1473-3099(18)30043-4
Firth, Cadhla, Andrew Kitchen, Beth Shapiro, Marc a. Suchard, Edward C. Holmes, and Andrew Rambaut. 2010. “Using Time-Structured Data to Estimate Evolutionary Rates of Double-Stranded DNA Viruses.” Molecular Biology and Evolution 27 (9): 2038–51.
Gigante, C. M., S. Smole, K. Wilkins, A. McCollum, C. Hutson, W. Davidson, A. Rao, C. Brown, and Y. Li. 2022. “Monkeypox Virus Isolate MPXV_USA_2022_MA001, Complete Genome.” NCBI Genbank. May 21, 2022. https://www.ncbi.nlm.nih.gov/nuccore/ON563414
Harris, Reuben S., Kate N. Bishop, Ann M. Sheehy, Heather M. Craig, Svend K. Petersen-Mahrt, Ian N. Watt, Michael S. Neuberger, and Michael H. Malim. 2003. “DNA Deamination Mediates Innate Immunity to Retroviral Infection.” Cell 113 (6): 803–9.
Isidro, Joana, Vítor Borges, Miguel Pinto, Rita Ferreira, Daniel Sobral, Alexandra Nunes, João Dourado Santos, Maria José Borrego, et al. 2022. “First Draft Genome Sequence of Monkeypox Virus Associated with the Suspected Multi-Country Outbreak, May 2022 (confirmed Case in Portugal).” Virological. May 19, 2022. https://pando.tools/t/799
Isidro, Joana, Vítor Borges, Miguel Pinto, Rita Ferreira, Daniel Sobral, Alexandra Nunes, João Dourado Santos, Verónica Mixão, et al. 2022. “Multi-Country Outbreak of Monkeypox Virus: Genetic Divergence and First Signs of Microevolution.” Virological. May 23, 2022. https://pando.tools/t/806
Kremer, Melanie, Yasemin Suezer, Yolanda Martinez-Fernandez, Carsten Münk, Gerd Sutter, and Barbara S. Schnierle. 2006. “Vaccinia Virus Replication Is Not Affected by APOBEC3 Family Members.” Virology Journal 3 (October): 86.
Likos, Anna M., Scott A. Sammons, Victoria A. Olson, A. Michael Frace, Yu Li, Melissa Olsen-Rasmussen, Whitni Davidson, et al. 2005. “A Tale of Two Clades: Monkeypox Viruses.” The Journal of General Virology 86 (Pt 10): 2661–72.
Mauldin, Matthew R., Andrea M. McCollum, Yoshinori J. Nakazawa, Anna Mandra, Erin R. Whitehouse, Whitni Davidson, Hui Zhao, et al. 2022. “Exportation of Monkeypox Virus From the African Continent.” The Journal of Infectious Diseases 225 (8): 1367–76.
Nakazawa, Yoshinori, Matthew R. Mauldin, Ginny L. Emerson, Mary G. Reynolds, R. Ryan Lash, Jinxin Gao, Hui Zhao, et al. 2015. “A Phylogeographic Investigation of African Monkeypox.” Viruses 7 (4): 2168–84.
Selhorst, Philippe, Antonio Mauro Rezende, Tessa de Block, Sandra Coppens, Hilde Smet, Joachim Mariën, Anne Hauner, et al. 2022. “Belgian Case of Monkeypox Virus Linked to Outbreak in Portugal.” Virological. May 20, 2022. https://pando.tools/t/801
Vanmechelen, Bert, Tony Wawina-Bokalanga, and Piet Maes. 2022. “A Monkeypox Virus Genome from a Second Belgian Case.” Virological. May 23, 2022. https://pando.tools/t/807
Yinka-Ogunleye, Adesola, Olusola Aruna, Mahmood Dalhat, Dimie Ogoina, Andrea McCollum, Yahyah Disu, Ibrahim Mamadu, et al. 2019. “Outbreak of Human Monkeypox in Nigeria in 2017-18: A Clinical and Epidemiological Report.” The Lancet Infectious Diseases 19 (8): 872–79.
Yong, S. E. F., O. T. Ng, Z. J. M. Ho, T. M. Mak, K. Marimuthu, S. Vasoo, T. W. Yeo, et al. 2020. “Imported Monkeypox, Singapore.” Emerging Infectious Diseases 26 (8). 10.3201/eid2608.191387
Yu, Qin, Renate König, Satish Pillai, Kristopher Chiles, Mary Kearney, Sarah Palmer, Douglas Richman, John M. Coffin, and Nathaniel R. Landau. 2004. “Single-Strand Specificity of APOBEC3G Accounts for Minus-Strand Deamination of the HIV Genome.” Nature Structural & Molecular Biology 11 (5): 435–42.
Zhang, Hui, Bin Yang, Roger J. Pomerantz, Chune Zhang, Shyamala C. Arunachalam, and Ling Gao. 2003. “The Cytidine Deaminase CEM15 Induces Hypermutation in Newly Synthesized HIV-1 DNA.” Nature 424 (6944): 94–98.