An updated version of this report is now available as part of a preprint at:
https://www.biorxiv.org/content/10.1101/2023.01.23.525187v1
Áine O’Toole & Andrew Rambaut
Institute of Ecology and Evolution
University of Edinburgh
2022-06-05We gratefully acknowledge the groups that have been publicly sharing MPXV genome sequence data which makes analyses such as this possible. These data are listed and cited in Table 1.
In a previous post we hypothesised that the current monkeypox virus (MPXV) epidemic had arisen from a non-human animal to human jump prior to 2017. This was based on the observation of a preponderance of a specific mode of mutation that is characteristic of cytosine deamination by molecules in the APOBEC3 family.
Here we show a linear accumulation of these mutation with time of collection of genome sequences, estimate the rate of accumulation and extrapolate back to estimate the time that these mutation started to accumulate. If we assume that APOBEC3-type mutations are indicative of replication within human cells and that all such mutations reconstructed in the phylogenetic tree of hMPXV1 genomes are the result of APOBEC3, then a simple extrapolation provides an estimate for the date of jump into the human population (Figure 1).
We estimate that APOBEC3-type mutations accumulate during human to human transmission at the rate of about 9 per year (8.74 with a 95% interval 7.34 – 10.15) which corresponds to an evolutionary rate of 4.43 × 10-5 substitutions per site per year (3.72 – 5.15 × 10-5) for a genome length of 197,210 nucleotides. This rate is approximately 20-fold greater than the long term evolutionary rate estimated for MPXV in the non-human animal reservoir of 1.93 × 10-6 (95% highest posterior density interval 1.21–2.66 × 10-6) (Patrono et al. 2020).
The estimate for the date of emergence of hMPXV1 in the human population is 5-Apr-2016 (95% highest posterior density intervals of 14-Jul-2015 – 18-Dec-2016).
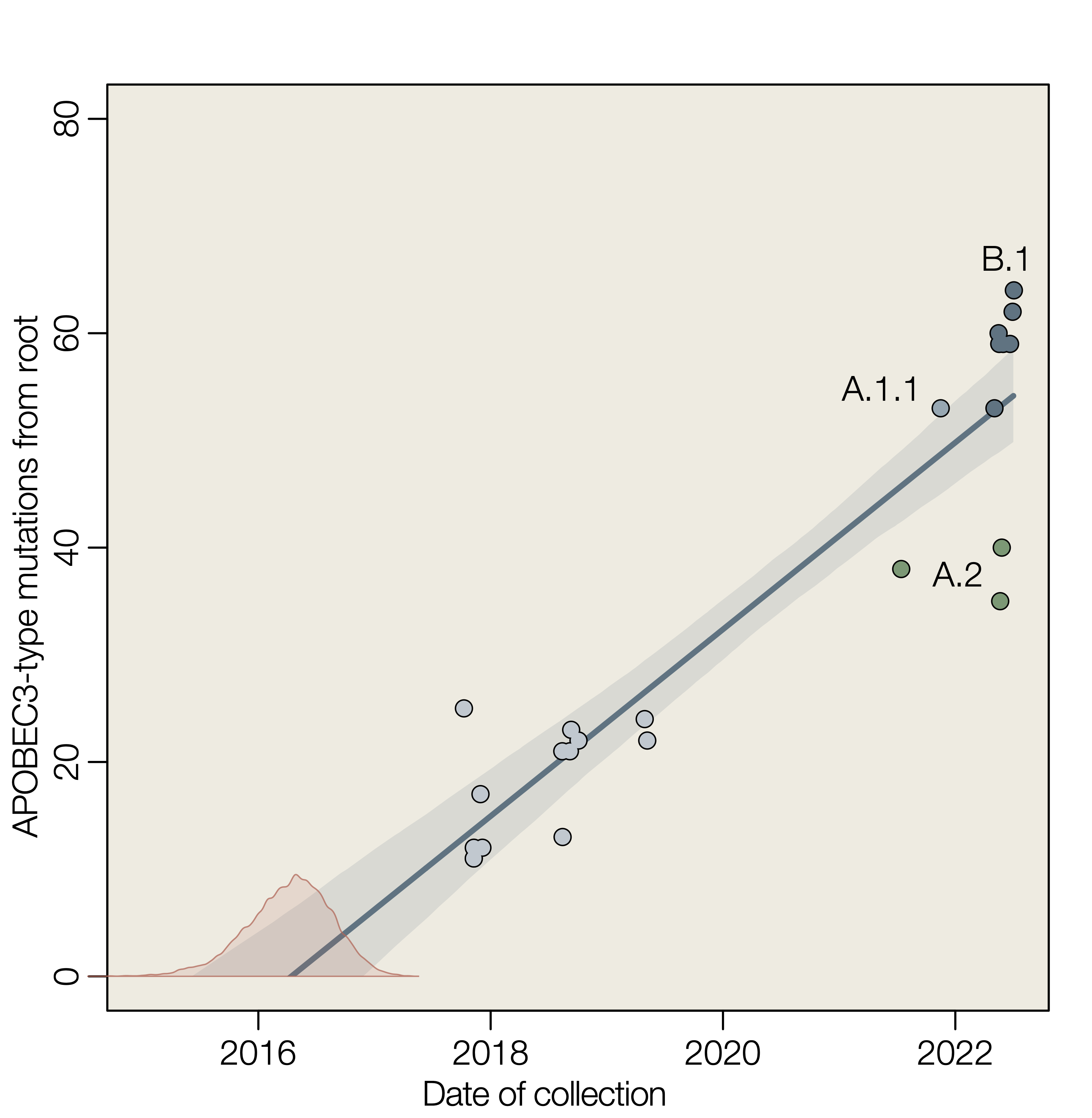
Figure 1 | Root to tip linear regression of APOBEC3-type mutations against time of collection. Slope is an estimate of the rate in substitutions per year (mean 8.74, 95% interval 7.34 – 10.15). Red density is an estimate of the time at y=0 (mean 2016.26, 95% highest posterior density interval 2015.54 – 2016.97).
Methods
The genomes used in this study are listed in Table 1. We arbitrarily selected 7 genomes from the B.1 lineage to span the time range for this lineage. The genome sequences were aligned to the Clade 3 reference genome (accession NC_063383) using minimap2 (Li 2018) and Squirrel. A maximum likelihood phylogenetic tree was constructed using IQTree2 (Minh et al. 2020) using KJ642617 (Nigeria, 1971) and KJ642615 (Nigeria, 1978) as outgroups (Figure 2). The putative ancestral genomes we reconstructed at each node using IQTree2 and then cumulative numbers of APOBEC3-type mutations (GA->AA or TC->TT dinucleotide transitions) were counted for each tip in the tree.
A linear model of APOBEC3-type mutations against date of collection was fitted using the quadratic approximation (quap function in the Rethinking package in R).
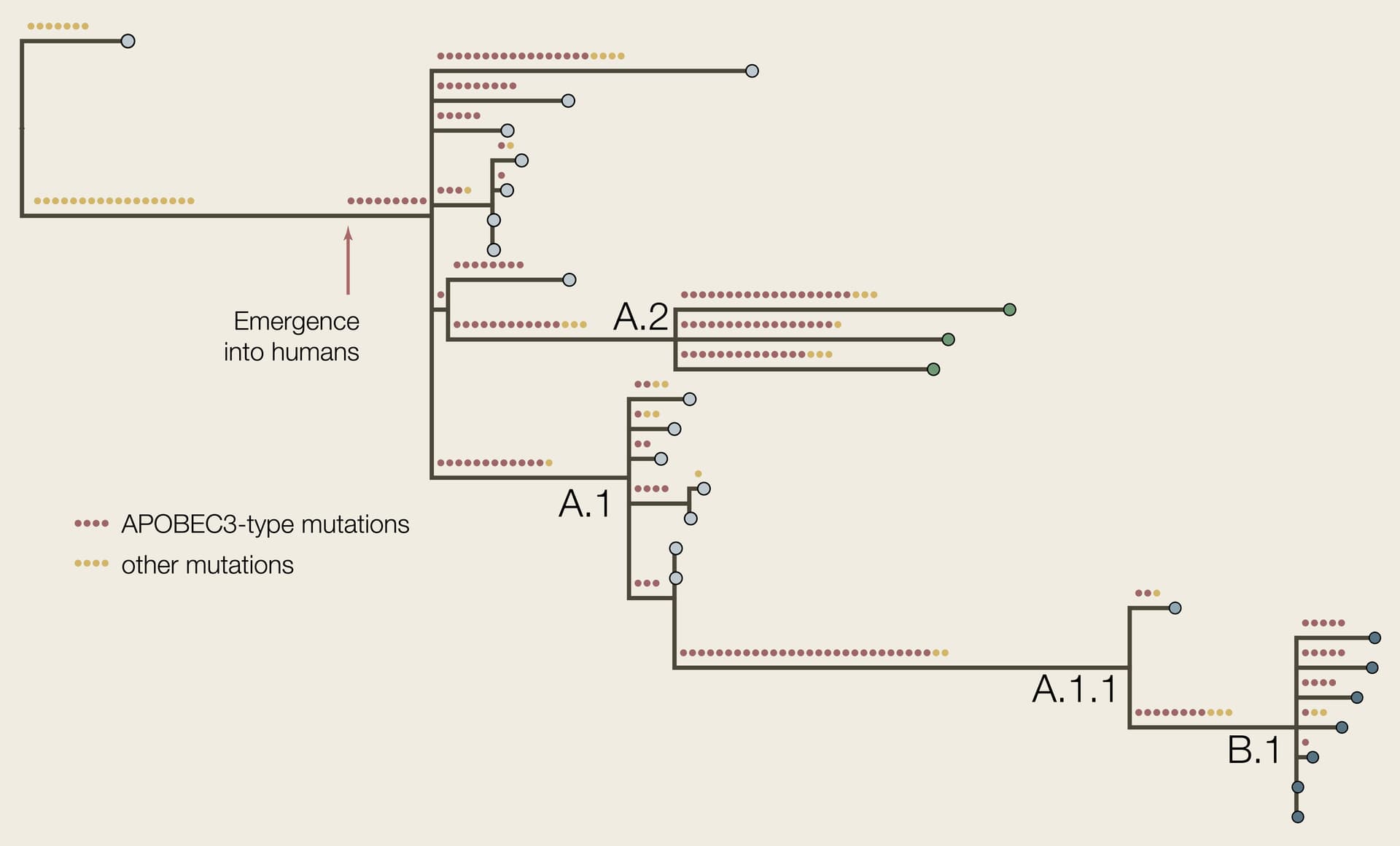
Figure 2 | The phylogenetic tree of the genomes listed here with APOBEC3-type and other mutations reconstructed. The red arrow represents the putative point at which the virus emerged into the human population if all APOBEC3-type mutations accrued in humans.
Caveats
The tips of the tree are not independent data because they share evolutionary history through common ancestry. Where genomes had known epidemiological linkage, only one representative was included. This effect can be seen in Figure 1 where the data points for particular lineages cluster together (e.g., A.1). This will tend to result in an underestimate of the uncertainty of the parameters.
C->T and G->A mutations are the most common so we would expect some APOBEC3-like mutations to occur by chance due to polymerase error alone (in a presumed non-human animal host). Thus the date of emergence should be considered an upper bound assuming all such mutations occurred after the jump to humans.
Table 1 | MPXV genome sequences used in this study.
| Accession | Name | Country | Year | Clade/Lineage | Reference |
|---|---|---|---|---|---|
| KJ642617 | Nigeria-SE-1971 | Clade 3 | Nigeria | 1971 | Nakazawa et al. 2015 |
| KJ642615 | W-Nigeria | Clade 3 | Nigeria | 1978 | Nakazawa et al. 2015 |
| MK783027 | 3018 | hMPXV1 A | Nigeria | 2017-11-09 | Yinka-Ogunleye et al. 2019 |
| MK783028 | 3019 | hMPXV1 A | Nigeria | 2017-11-09 | Yinka-Ogunleye et al. 2019 |
| MK783029 | 3029 | hMPXV1 A | Nigeria | 2017-12-06 | Yinka-Ogunleye et al. 2019 |
| MK783030 | 3025 | hMPXV1 A | Nigeria | 2017-11-30 | Yinka-Ogunleye et al. 2019 |
| MK783031 | 3020 | hMPXV1 A | Nigeria | 2017-11-09 | Yinka-Ogunleye et al. 2019 |
| MK783032 | 3030 | hMPXV1 A | Nigeria | 2017-11-01 | Yinka-Ogunleye et al. 2019 |
| MK783033 | 2920 | hMPXV1 A | Nigeria | 2017-10-09 | Yinka-Ogunleye et al. 2019 |
| NC_063383 | M5312_HM12 | hMPXV1 A | Nigeria | 2018-08-15 | Mauldin et al., 2022 |
| MT903341 | M5320_M15 | hMPXV1 A.1 | Nigeria | 2018-08-14 | Mauldin et al., 2022 |
| MT903342 | Singapore | hMPXV1 A.1 | Singapore | 2019-04-30 | Mauldin et al., 2022 |
| MT903343 | UK_P1 | hMPXV1 A.1 | UK | 2018-09-07 | Mauldin et al., 2022 |
| MT903344 | UK_P2 | hMPXV1 A.1 | UK | 2018-09-11 | Mauldin et al., 2022 |
| MT903345 | UK_P3 | hMPXV1 A.1 | UK | 2018-09-22 | Mauldin et al., 2022 |
| MN648051 | Israel | hMPXV1 A.1 | Israel | 2018-10-04 | Cohen-Gihon et al. 2020 |
| MT250197 | Singapore_2019 | hMPXV1 A.1 | Singapore | 2019-05-08 | Yong et al. 2020 |
| ON676708 | MD001 | hMPXV1 A.1.1 | USA | 2021-11-16 | Gigante et al. 2022 |
| ON674051 | FL001 | hMPXV1 A.2.1 | USA | 2022-05-22 | Gigante et al. 2022 |
| ON676707 | TX001 | hMPXV1 A.2 | USA | 2021-07-15 | Gigante et al. 2022 |
| ON675438 | VA001 | hMPXV1 A.2 | USA | 2022-05-27 | Gigante et al. 2022 |
| ON563414 | MA001 | hMPXV1 B.1 | USA | 2022-05-19 | Gigante et al. 2022 |
| ON803417 | UN-NML-2836 | hMPXV1 B.1 | Canada | 2022-05-17 | Knox et al. 2022 |
| ON880542 | UN-NML-3348 | hMPXV1 B.1 | Canada | 2022-05-31 | Duggan et al. 2022 |
| ON959143 | MPX-96 | hMPXV1 B.1 | Finland | 2022-06-16 | Kant et al. 2022 |
| OP062230 | UN-NML-3864 | hMPXV1 B.1 | Canada | 2022-06-22 | Duggan et al. 2022 |
| OP062235 | UN-NML-3934 | hMPXV1 B.1 | Canada | 2022-06-30 | Duggan et al. 2022 |
| OX248696 | RNA | hMPXV1 B.1 | Slovakia | 2022-07-04 | Comenius University of Bratislava, Ilkovicova 8, 841 04 Bratislava, Slovakia |
References
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, Lanfear R (2020) IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol., 37:1530-1534. DOI: 10.1093/molbev/msaa015
Cohen-Gihon, Inbar, Ofir Israeli, Ohad Shifman, Noam Erez, Sharon Melamed, Nir Paran, Adi Beth-Din, and Anat Zvi. 2020. “Identification and Whole-Genome Sequencing of a Monkeypox Virus Strain Isolated in Israel.” Microbiology Resource Announcements 9 (10). DOI: 10.1128/MRA.01524-19
Duggan,A., Hole,D., Knox,N., Yadav,C., Haidl,E., Chapel,M., Domselaar,G.V., Fernando,L., Graham,M., Antonation,K., Audet,J., Hagan,M., Safronetz,D., Leung,A., Peters,G., Go,A., Laminman,V., Kaplen,B., Jolly,G., Charest,H., Levade,I. and Fafard,J. (2022) Public Health Agency of Canada, National Microbiology Laboratory, 1015 Arlington Street, Winnipeg, MB R3E 3R2, Canada
Gigante,C.M., Myers,R., Seabolt,M.H., Wilkins,K., McCollum,A., Hutson,C., Davidson,W., Rao,A., Blythe,D. and Li,Y. Division of High-Consequence Pathogens and Pathology, Centers for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30329, USA
Isidro, J., Borges, V., Pinto, M. et al. Phylogenomic characterization and signs of microevolution in the 2022 multi-country outbreak of monkeypox virus. Nat Med (2022). DOI:10.1038/s41591-022-01907-y
Kant,R., Smura,T., Vauhkonen,H. and Vapalahti,O.(2022) Department of Virology, Faculty of Medicine, University of Helsinki, Hartmaninkatu 3, Helsinki, Helsinki 00014, Finland
Knox,N., Duggan,A., Yadav,C., Hole,D., Haidl,E., Chapel,M., Jolly,G., Domselaar,G.V., Antonation,K., Leung,A., Fernando,L., Audet,J., Hagan,M., Graham,M., Griffiths,E., Safronetz,D., Charest,H., Levade,I. and Fafard,J. (2022) Public Health Agency of Canada, National Microbiology Laboratory, 1015 Arlington Street, Winnipeg, MB R3E 3R2, Canada
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics, 34:3094-3100. DOI: 10.1093/bioinformatics/bty191
Likos, Anna M., Scott A. Sammons, Victoria A. Olson, A. Michael Frace, Yu Li, Melissa Olsen-Rasmussen, Whitni Davidson, et al. 2005. “A Tale of Two Clades: Monkeypox Viruses.” The Journal of General Virology 86 (Pt 10): 2661–72.
Mauldin, Matthew R., Andrea M. McCollum, Yoshinori J. Nakazawa, Anna Mandra, Erin R. Whitehouse, Whitni Davidson, Hui Zhao, et al. 2022. “Exportation of Monkeypox Virus From the African Continent.” The Journal of Infectious Diseases 225 (8): 1367–76. DOI:10.1093/infdis/jiaa559
Nakazawa, Yoshinori, Matthew R. Mauldin, Ginny L. Emerson, Mary G. Reynolds, R. Ryan Lash, Jinxin Gao, Hui Zhao, et al. 2015. “A Phylogeographic Investigation of African Monkeypox.” Viruses 7 (4): 2168–84. DOI:10.3390/v7042168
Patrono, L.V., Pléh, K., Samuni, L. et al. (2020) Monkeypox virus emergence in wild chimpanzees reveals distinct clinical outcomes and viral diversity. Nat Microbiol 5, 955–965. DOI: 10.1038/s41564-020-0706-0
Yinka-Ogunleye, Adesola, Olusola Aruna, Mahmood Dalhat, Dimie Ogoina, Andrea McCollum, Yahyah Disu, Ibrahim Mamadu, et al. 2019. “Outbreak of Human Monkeypox in Nigeria in 2017-18: A Clinical and Epidemiological Report.” The Lancet Infectious Diseases 19 (8): 872–79. DOI:10.1016/s1473-3099(19)30294-4
Yong, S. E. F., O. T. Ng, Z. J. M. Ho, T. M. Mak, K. Marimuthu, S. Vasoo, T. W. Yeo, et al. 2020. “Imported Monkeypox, Singapore.” Emerging Infectious Diseases 26 (8). DOI:10.3201/eid2608.191387