Gregory Destras1,2,3, Antonin Bal1,2,3, Bruno Simon1,2, Quentin Semanas1,2, Hadrien Règue1,2, Bruno Lina1,2,3, Laurence Josset1,2,3
- GenEPII Sequencing Platform, Institut des Agents Infectieux, Hospices Civils de Lyon, F-69004, Lyon, France
- Laboratoire de Virologie, Institut des Agents Infectieux, Laboratoire associé au Centre National de Référence des virus des infections respiratoires, Hospices
Civils de Lyon, Lyon, France - CIRI, Centre International de Recherche en Infectiologie, Team VirPath, Univ Lyon, Inserm, U1111, Université Claude Bernard Lyon 1, Lyon, France
Whole-genome sequencing (WGS) of the first cases of Monkeypox virus responsible of the 2022 outbreak in Europe has shown that sequences were highly similar and belonged to the West African clade [https://pando.tools/t/first-monkeypox-genome-sequence-from-the-netherlands/821; https://pando.tools/t/multi-country-outbreak-of-monkeypox-virus-genetic-divergence-and-first-signs-of-microevolution/806]. Most of these sequences were initially performed using ONT. Obtaining Monkeypox virus sequences from different platform technologies is crucial to avoid sequencing errors.
Here we report the first french Monkeypox whole-genome sequence performed on Illumina platform, from a male patient returning from Canary Islands on 19 May 2022. Skin lesions were sampled on 22 May 2022 at Emergency Unit of Hospices Civils de Lyon, France
Metagenomic next-generation sequencing was performed at GenEPII sequencing platform of Hospices Civils de Lyon. Total nucleic acid extraction was performed on Emag platform (Biomerieux, Marcy l’Etoile, France) and eluted into 50µL. DNA concentration was quantified at 0.09 ng/µL using the Qubit dsDNA HS Assay Kit 2.0 (Thermo Fisher Scientific, Dreieich, Germany). Direct DNA libraries were carried out in quadruplicate from 2µL using DNAprep kit (Illumina, San Diego, USA) and according to the manufacturer’s recommendation number of cycles (12 cycles for low-input). The fragment size was evaluated around 338bp on a 4150 TapeStation Instrument (Agilent, Santa Clara, USA) and DNA library concentration was quantified at 4.10ng/µL. Sequencing was then achieved using a SP 1.5 2x100bp cartridge on NovaSeq instrument (Illumina, San Diego, CA, USA).
A total of 84M paired-end reads was obtained per sample. Analysis was performed using our in-house pipeline developed during COVID-19 pandemics (GitHub - genepii/seqmet). Reads were mapped against the Monkeypox reference genome (MPXV_UK_P1, MT903343.1).
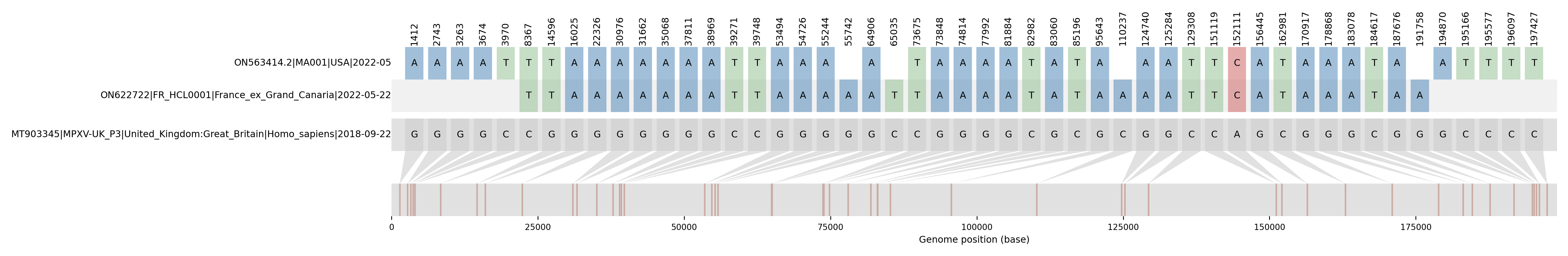
We obtained 4 full genomes (950X mean depth for each sample), each characterized by a drop in the coverage depth distribution corresponding to a deletion of a repeated 10 nt motif (CAATCTTTCT) at position 133166 in a non-coding region (Figure 1). There was 0 nucleotide difference among the 197.378 bp length of the four genomes (Figure 2). These results suggest that this protocol is reproducible after DNA extraction. A consensus sequence has been deposited on GenBank (ON622722.2, updated on 2022-06-02) and available here.
Updated genome on 2022-06-02: IlluminaFranceMPXV_FR_HCL0001_2022ON6227222022-05-22.zip (56.0 KB)
This virus carries 56 SNPs and 3 deletions, including two in homopolymeric regions in comparison with the reference genome.

Figure 1. Coverage depth distributions of the MPXV genome performed in quadruplicate.

Figure 2. Distance matrix between Monkeypox viral sequences performed in quadruplicate and the reference genome MPXV_UK_P1.
The 56 SNPs and 3 deletions compared to the reference correspond to: Δ599-600delTT; G1267A ; G2596A ; G3116A ; G3527A ; C3823T ; C7776T ; G14005T ; G15433A ; G21728A ; G25666A ; A28843G ; G30372A ; G31058A ; G34464A ; G37207A ; G38365A ; C38667T ; C39124T ; C39144T ; G52890A ; G54122A ; G54640A ; G55138A ; G64302A ; C64431T ; C73071T ; G73244A ; G74210A ; G77388A ; G81280A ; C82378T ; G82456A ; C84592T ; A92354G ; G95039A ; C109632A ; G124135A ; G124679A ; C128703T ; Δ133166-133178delCAATCTTTCT ; C150474T ; A151466C ; G155800A ; G162248A ; C162336T ; G170267A ; G178150A ; G181900A ; C183439T; G186498A ; A188414G ; G190580A ; G193312A ; C193608T ; C194019T ; C194539T ; C195868T ; Δ196521-196522delAA
Acknowledgements
We would like to thank all members of the GenEPII sequencing platform who contributed to the sequencing of this case. We also thank the members of the emergency unit, the infectious disease unit and the virology laboratory of Hospices Civils de Lyon who contributed to this investigation. This work was carried out within the framework of the French consortium on surveillance and research on infections with emerging pathogens via microbial genomics (consortium relatif à la surveillance et à la recherche sur les infections à pathogens EMERgents via la GENomique microbienne EMERGEN; Consortium EMERGEN)
Revised version on 2022-06-02