Identification of a novel SARS-CoV-2 Spike 69-70 deletion lineage circulating in the United States
Brendan B. Larsen, Michael Worobey
Department of Ecology and Evolutionary Biology, University of Arizona
New mutations are continually emerging in SARS-CoV-2 that could impact viral phenotype. Genome sequencing of SARS-CoV-2 is critical for identifying and tracking these mutations. Recently, a novel lineage was identified in the UK (named lineage B.1.1.7) that exhibits a number of mutations that could impact its spread1.
One of the mutations in the B.1.1.7 lineage is an in-frame, 6bp deletion at Spike amino acid positions 69 and 70 (69-70del). This particular deletion has been observed in multiple distinct lineages besides B.1.1.7, notably in the mink cluster V lineage from Denmark and at low levels elsewhere in the world2. This deletion is frequently observed with other Spike amino acid changes. Some evidence suggests this deletion alone might make the virus more transmissible, but in vivo evidence for increased transmission for this particular deletion is still absent.
Here, we identify a novel lineage circulating in the United States that contains a similar deletion from sequences downloaded from GISAID as of 12/30/2020. In total this lineage is made up of twelve genomes from six states collected from 10/21 to 12/18. There is one additional genome from Ecuador that nests within the US sequences (EPI_ISL_672005) that does not contain a collection date and therefore was not included in further analyses.
In order to determine if sequences with the 69-70del in the United States represent a single deletion event and not multiple independent events, all genomes deposited to GISAID were downloaded and sequences with the 20C clade defining mutation C1059T were pulled out to make alignment and phylogeny inference more manageable. The twelve genomes with the 69-70del are monophyletic with respect to all other genomes in GISAID, and share a common ancestor with other sequences collected in the United States (Figure 1). The 69-70del occurs in a nested subset of pangolin lineage B.1.346 (12/133 genomes in GISAID).
Interestingly, this lineage only contains 2 additional nonsynonymous mutations in spike aside from the 69-70 deletion. All 12 genomes contain the canonical spike mutation D614G. A further six genomes from Utah in December contain a C1236S mutation. BEAST analysis using tip dates and a strict clock prior of 8e-4 infers an average time of the most recent common ancestor estimate of 9/11/2020, suggesting this variant has been circulating at very low levels in the United States for at least the past 4 months (Figure 2). Although it has spread to multiple states, this lineage remains at very low levels compared to the total number of genomes sequenced from the United States over this time period, suggesting it is not spreading rapidly at this point (12 genomes out of 8,170 sequenced in the United States over this time period).
The S-dropout is being used by public health officials to identify sequences with the 69-70del since the deletion occurs where the qPCR probe binds. Most notably, this has been used to screen for B.1.1.7 in the UK and in the United States. As others have already noted on social media, such as Nate Grubaugh, the presence of this novel lineage suggests that an S-dropout result in the United States is not necessarily B.1.1.7 since there are a low level of viruses with this mutation also circulating. Full genome sequencing is needed to confirm the presence of B.1.1.7.
Although the full significance of this deletion remains to be determined, due to the independent emergence of this deletion across the world, and in vitro studies suggesting it may be more transmissible or may impact immune escape, this particular lineage should be further monitored.

Figure 1. Maximum likelihood phylogeny of the novel 69-70del lineage (in red) with its closest relatives. The phylogeny is based on all genomes in GISAID downloaded on 12/30/2020 and inferred with IQTREE 2.0.
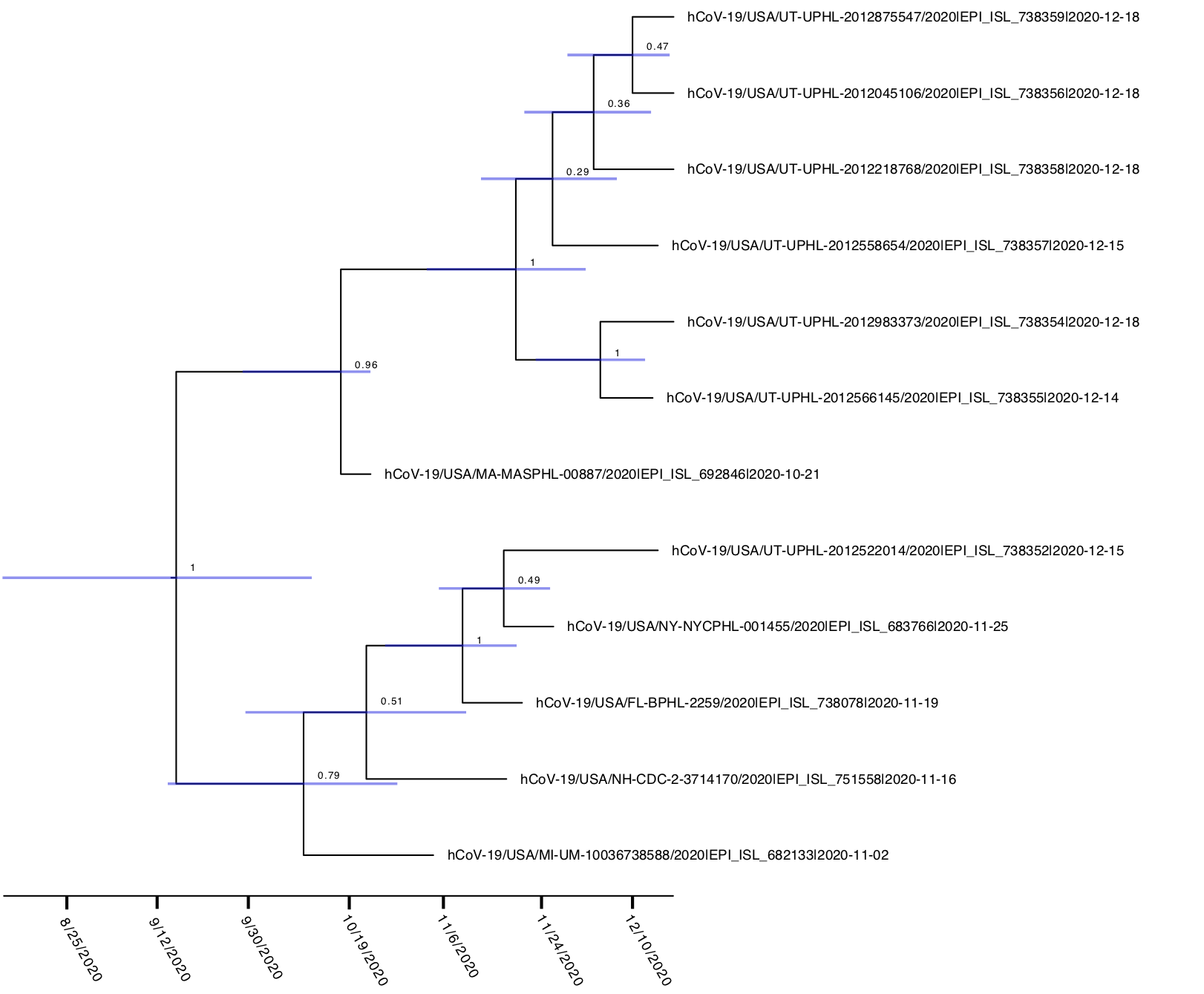
Figure 2. BEAST time calibrated phylogeny of the novel 69-70 del lineage. Numbers at nodes represent posterior probabilities and blue bars represent 95% HPD estimates for the timing of each node.
Acknowledgements
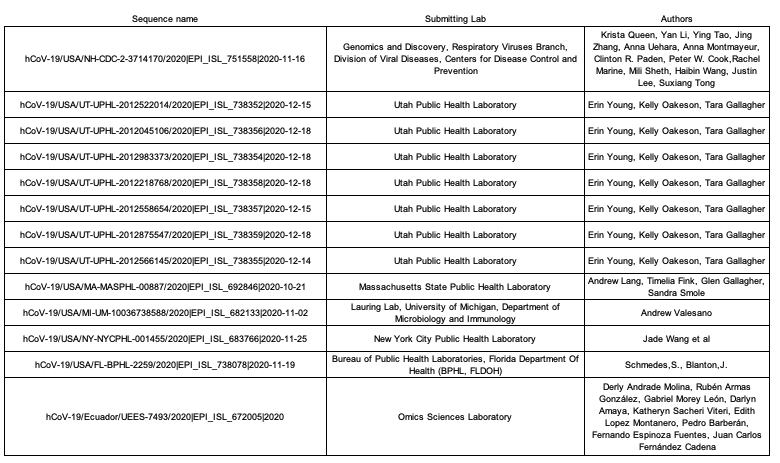
We gratefully acknowledge the laboratories and researchers who made these SARS-CoV-2 genomes available on GISAID:Utah Public Health Laboratory, Florida Department Of Health, Massachusetts State Public Health Laboratory, New York City Public Health Laboratory, University of Michigan Clinical Microbiology Laboratory, and the Centers for Disease Control and Prevention
References
- Preliminary analysis of SARS-CoV-2 importation & establishment of UK transmission lineages. Virological https://pando.tools/t/preliminary-analysis-of-sars-cov-2-importation-establishment-of-uk-transmission-lineages/507 (2020).
- Kemp, S. A. et al. Recurrent emergence and transmission of a SARS-CoV-2 Spike deletion ΔH69/V70. Cold Spring Harbor Laboratory 2020.12.14.422555 (2020) doi:10.1101/2020.12.14.422555.