Resurgence of Ebola virus in Guinea after 5 years calls for careful attention to survivors without creating further stigmatization
Alpha K. Keita1,2, Ariane Düx3, Haby Diallo1, Sébastien Calvignac-Spencer3, Mamadou S Sow2,4, Mamadou Bhoye Keita5, Youssouf Sidibe6, Ahidjo Ayouba2, Frédéric Lemarcis1,2, Almudena Mari Saez3, Mandiou Diakite7, Kaba Kourama5, Ibrahima Camara1, Eric Delaporte2, Fabian Leendertz3, Martine Peeters2, Abdoulaye Toure1,5
- Centre de Recherche et de Formation en Infectiologie de Guinée (CERFIG), Université de Conakry, Guinea
- TransVIHMI, Montpellier University/IRD/INSERM, Montpellier, France
- Robert Koch Institute, Berlin, Germany
- Hôpital National Donka, Service des Maladies Infectieuses et Tropicales, Conakry, Guinea
- Institut National de Santé Publique de Guinée, Conakry, Guinea
- Laboratoire de Biologie médicale, Hôpital Régional de Nzérékoré, Nzérékoré, Guinea
- Direction Nationale des Laboratoires, Ministère de la Santé, Conakry, Guinea
Seven years after the declaration of the first Ebola virus disease (EVD) epidemic in Guinea, the country is facing a new Ebola outbreak in a town located only 200 km from the epicenter of the previous epidemic. In order to discriminate whether this new outbreak was caused by a new zoonotic transmission event or by resurgence of the viral strain that circulated in the previous West African outbreak whole genome sequencing was attempted at CERFIG in Conakry, Guinea, on viral extracts from samples that were positive for EBOV (Zaire ebolavirus) NP and GP on the GeneXpert molecular diagnostic platform (Xpert Ebola Assay).
For this, we generated Illumina libraries which were enriched in filoviral sequences by hybridization capture before their sequencing on an Illumina iSeq platform. We used the resulting reads to generate consensus sequences, applying a conservative 20X, 90% agreement base calling threshold. This allowed us to determine four genomic sequences covering ≥99.9% of the EBOV reference genome at a depth of 223-763X (unique reads). Three of the genomes were strictly identical and the fourth only exhibited a single nucleotide substitution to the others.
We aligned these genomes and a selection of representatives from earlier outbreaks using MAFFT and then used IQ-TREE to estimate a maximum likelihood phylogenetic tree from this alignment. In the resulting tree, the new Guinean EBOV genomes nest within the clade formed by sequences from the first epidemic, within which they also form a well-supported sub-clade with genomes from Guinea, Liberia and Sierra Leone from the previous outbreak (2013-2016) (Figure 1). This pattern shows clearly that the new outbreak is the result of the resurgence of a strain that previously circulated in the West African outbreak from 2013-2016. The unexpectedly short branch leading to the 2021 Guinean EBOV genomes suggests a marked substitution rate slowdown, which might be due to latency in a survivor.
The human origin of the current EVD outbreak and the associated shift in our perception of EBOV emergence, call for careful attention to survivors. The possibility of resurgence of Ebola, up to 5 years or more after EVD, opens new challenges for survivors, their families and their communities but also for the health system that has to create ways to work with communities, known and unknown survivors, without creating further stigmatization.
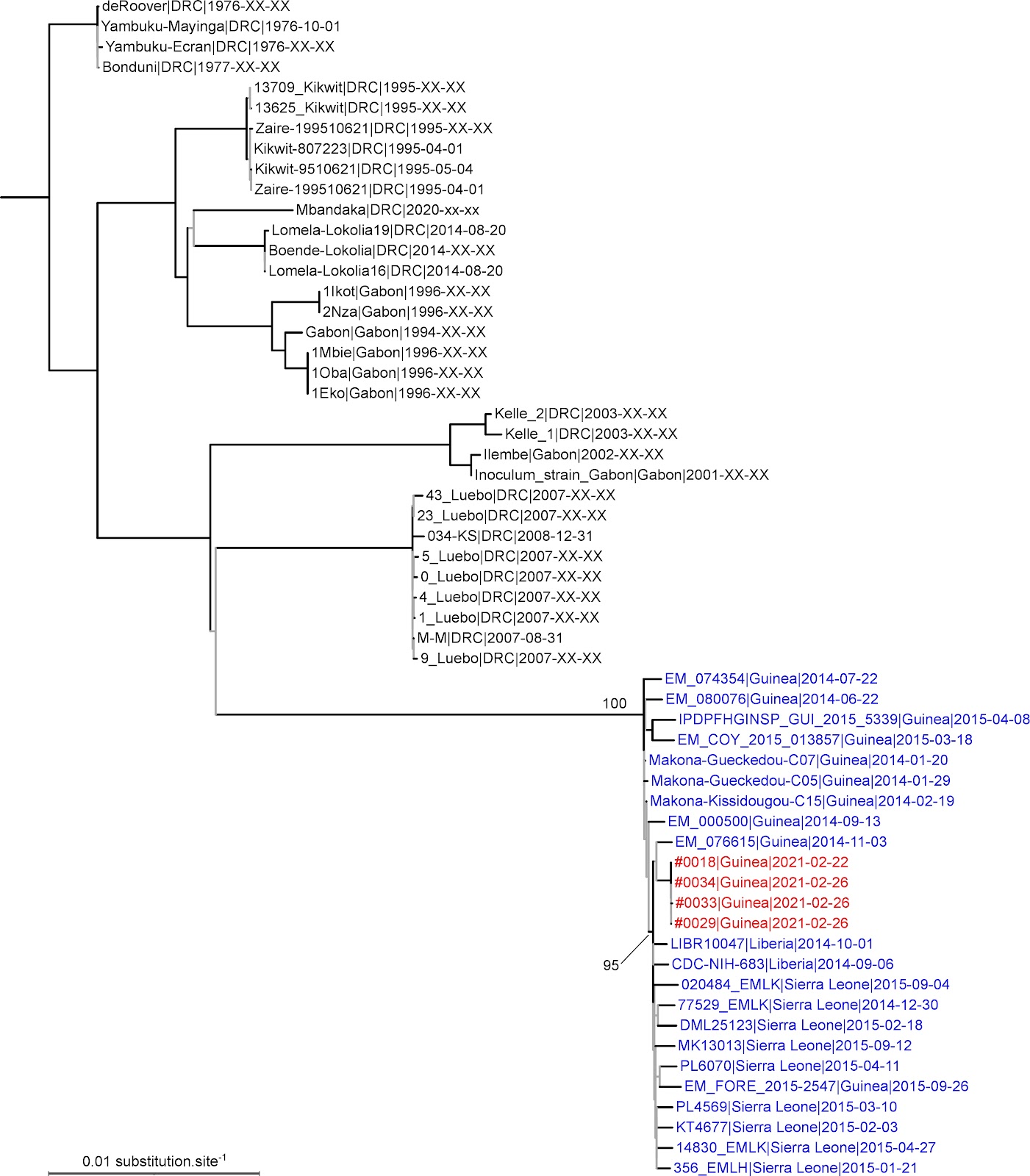
Figure 1. Maximum likelihood tree derived from a whole genome alignment of EBOV sequences. Tip labels corresponding to EBOV genomes from the 2013-6 West African outbreak are blue; EBOV genomes from the 2021 Guinean outbreak red. Branches with SH-like aLRT support < 0.95 are grey. By convention, the tree was rooted on the branch leading to the clade formed by late 1970s EBOV genomes.