Genomic surveillance of SARS-CoV-2 reveals community transmission of a major lineage during the early pandemic phase in Brazil
Paola Cristina Resende 1a, Edson Delatorre 2, Tiago Gräf 3, Daiana Mir 4, Fernando do Couto Motta 1, Luciana Reis Appolinario 1, Anna Carolina Dias da Paixão 1, Maria Orgzwalska 1, Braulia Caetano 1, Sandra Bianchini Fernandes 5, Lucas A Vianna 6, Jean F G Ferro 7, Larissa da Costa Souza 8, Leandro Ferraz 9, Julio Croda 10,11, André Abreu 12, Gonzalo Bello 13a*, Marilda M Siqueira 1*
*Both authors contributed equally to this work.
a Correspondent authors
Paola Cristina Resende - [email protected]
Gonzalo Bello - [email protected]
Filiations
1 Laboratory of Respiratory Viruses and Measles, Oswaldo Cruz Institute (IOC), FIOCRUZ, Rio de Janeiro, Brazil. SARS-CoV-2 National Reference Laboratory for the Brazilian Ministry of Health (MoH) and Regional Reference Laboratory in Americas for the Pan-American Health Organization (PAHO/WHO).
2 Departamento de Biologia. Centro de Ciências Exatas, Naturais e da Saúde, Universidade Federal do Espírito Santo, Alegre, Brazil.
3 Instituto Gonçalo Moniz, Fundação Oswaldo Cruz, Salvador, Brazil.
4 Unidad de Genómica y Bioinformática, Centro Universitario Regional del Litoral Norte, Universidad de la República, Salto, Uruguay.
5 Laboratório Central de Saúde Pública do Estado de Santa Catarina (LACEN-SC), Florianópolis, Santa Catarina, Brazil.
6 Laboratório Central de Saúde Pública do Estado Espírito Santo (LACEN-ES). Vitória, ES, Brazil.
7 Laboratório Central de Saúde Pública do Distrito Federal (LACEN-DF). Brasilia, DF, Brazil.
8 Laboratório Central de Saúde Pública de Alagoas (LACEN-AL). Maceió, AL, Brazil.
9 Laboratório Central de Saúde Pública da Bahia (LACEN-BA). Salvador, BA, Brazil.
10 Fiocruz Mato Grosso do Sul, Campo Grande, MS, Brazil.
11 Universidade Federal de Mato Grosso do Sul – UFMS, Campo Grande, MS, Brazil.
12 Coordenadoria Geral de Laboratórios - Brazilian Ministry of Health, Brasilia, Brazil.
13 Laboratório de AIDS e Imunologia Molecular, Instituto Oswaldo Cruz, Fundação Oswaldo Cruz, Rio de Janeiro, Brazil.
Introduction
COVID-19, disease caused by Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2), is leading high rates of acute respiratory syndrome, hospitalization and death ( 1, 2 ). Second in number of reported cases in the world, Brazil has reported 772.416 cases and 39.680 deaths (last update 10th June, 2020) ( 3 ). The first positive case of SARS-CoV-2 infection in Brazil was reported on 26th February, 2020 in Sao Paulo metropolitan region ( 4 ).
The rapid worldwide SARS-CoV-2 genomic surveillance response sharing via GISAID (https://www.gisaid.org/) genomic sequence and patient’s metadata, enabled fast dissemination of results and provided a view of early transmission patterns in this pandemic. The SARS-CoV-2 has diversified in several phylogenetic lineages while it spread geographically across the world ( 5-7 ). A SARS-CoV-2 lineage previously designated as “G” or “B1” clade was initially tracked as the most common variant in Europe and is currently one of the most active virus lineages in Europe and North America ( 5-7 ). Inspection of SARS-CoV-2 genome sequences from South America available on GISAID, revealed that most strains (82%) belong to the B.1 clade indicating that this lineage is also the most prevalent SARS-CoV-2 variant circulating in South America (Table 1).
Genomic epidemiology is a useful tool to track the community transmission of SARS-CoV-2 in different geographical settings. Previous studies revealed that SARS-CoV-2 epidemics in Australia ( 8, 9 ), Belgium ( 10 ), Denmark ( 11 ), France ( 12 ), Iceland ( 13 ), Israel ( 14 ), Netherlands ( 15 ) Spain ( 16 ) and the US ( 17-19 ), resulted from multiple independent introductions, followed by community dissemination of some viral strains that resulted in the emergence of national (or local) transmission clusters. Genetic analyses of 46 SARS-CoV-2 complete genomes from Brazil, 40 from the state of Minas Gerais ( 20 ) and six from the state of Sao Paulo ( 21 ), showed multiple independent importations and limited local spread during the initial stage of SARS-CoV-2 transmission in Brazil. Although some B.1 Brazilian sequences grouped together in small clades, these studies failed to detect transmission clusters of large size in Brazil ( 20 ). The SARS-CoV-2 genomes analyzed in previous studies were mostly recovered from individuals returning from international travel, rather than linked to community transmission, and thus might had not recovered the genetic diversity of SARS-CoV-2 strains spreading locally in Brazil.
Results
To investigate the SARS-CoV-2 strains responsible for community transmission in Brazil, we analyzed 81 new viral whole-genomes collected between 29th February and 28th April 2020, recovered from individuals that reside in the Brazilian states of Rio de Janeiro (n = 70), Distrito Federal (n = 5), Bahia (n = 2), Santa Catarina (n = 2), Alagoas (n = 1) and Espírito Santo (n = 1). Viral genomes were obtained from nasopharyngeal swabs from individuals with confirmed SARS-CoV-2 infection, mostly (91.1%) reporting no international travel (Supplementary table 1), who underwent testing and genomic sequencing at the Laboratory of Respiratory Viruses and Measles, Oswaldo Cruz Institute (IOC), FIOCRUZ, in Rio de Janeiro, Brazil ( 22 ). New Brazilian genome sequences of SARS-CoV-2 were assigned to viral lineages according to the nomenclature proposed by Rambaut et al (7) , using the pangolin web application (https://pangolin.cog-uk.io). Most (94%) Brazilian SARS-CoV-2 sequences here obtained were assigned to the B.1 clade, and particularly to the B.1.1 sub-clade (Table 1). The prevalence of the B.1.1 sub-clade in our dataset (90%) was much higher than that estimated from other Brazilian sequences (46%) available in GISAID.
Table 1. Prevalence of SARS-CoV-2 lineage B.1 and B.1.1 across South American countries.
| Country | Source | N | Lineage B.1 | Lineage B.1.1 |
|---|---|---|---|---|
| Brazil | This study | 81 (100%) | 76 (94%) | 73 (90%) |
| Brazil | GISAID | 89 (100%) | 76 (85%) | 41 (46%) |
| Argentina | GISAID | 29 (100%) | 29 (100%) | 16 (55%) |
| Chile | GISAID | 153 (100%) | 127 (83%) | 35 (23%) |
| Colombia | GISAID | 126 (100%) | 114 (90%) | 16 (14%) |
| Uruguay | GISAID | 45 (100%) | 16 (36%) | 7 (16%) |
We next download all SARS-CoV-2 B.1.1 complete genome sequences ( > 29 Kilobases) with appropriate metadata available in GISAID (https://www.gisaid.org/) as of 4th June. After excluding low-quality genomes ( > 10% of N) and nearly identical sequences (genetic similarity > 99.99%) sequences from the most densely sampled location (United Kingdom [UK]), we obtained a global reference B.1.1 dataset containing 3,764 sequences that were aligned with the new 73 B.1.1 Brazilian sequences identified in this study and subjected to maximum-likelihood phylogenetic analyses using IQTree v1.6.12 ( 23 ). Brazilian isolates were distributed throughout the phylogenetic tree, consistent with multiple independent introductions from abroad (Figure 1). A significant proportion of Brazilian B.1.1 sequences (65%, n = 74), however, branched in a well-supported (bootstrap [BP] = 74%) monophyletic lineage here designated as B.1.1.BR, which was nested within a larger and highly supported (BP = 87%) lineage containing basal sequences from Western Europe and Brazil, here referred as B.1.1.EU/BR (Figure 1).
The paraphyletic basal clade B.1.1.EU/BR comprises sequences from the UK, Switzerland, Netherlands, Australia and from the Brazilian states of Minas Gerais and Distrito Federal (Tables 2 and 3). In addition to sharing the three nucleotide mutations that define the lineage B.1.1 (G28881A, G28882A, G28883C), sequences from this clade also harbor a non-synonymous T29148C mutation at the Nucleocapsid protein (I292T). The monophyletic lineage B.1.1.BR comprise sequences from different Brazilian states (particularly from Rio de Janeiro, but also from Acre, Amapá, Distrito Federal, Maranhão and Pará), as well as a few sequences from South America (Argentina, Chile and Uruguay), North America (Canada and USA), Australia and England (Tables 2 and 3). Besides the mutation T29148C, all sequences of the B.1.1-BR cluster harbor the non-synonymous mutation T27299C at the ORF6 (I33T). Mutations T29148C or T27299C were not detected in the other 7,520 B.1.1 genomes with fully resolved nucleotides at those positions available in GISAID as of 6th June, supporting the hypothesis that they are synapomorphic traits of the B.1.1.EU/BR and B.1.1.BR clades, respectively.
Table 2. Prevalence of SARS-CoV-2 lineages B.1.1.EU/BR and B.1.1.BR across countries.
| Region | Country | Total SARS-Cov-2 | Lineage B.1.1.EU/BR | Lineage B.1.1.BR |
|---|---|---|---|---|
| South America | Brazil | 171 | 7 (4%) | 74 (43%) |
| Argentina | 29 | - | 4 (14%) | |
| Chile | 153 | - | 7 (5%) | |
| Uruguay | 45 | - | 1 (2%) | |
| North America | Canada | 227 | - | 1 (<1%) |
| US | 7,605 | - | 10 (<1%) | |
| Oceania | Australia | 1,899 | 1 (<1%) | 4 (<1%) |
| Europe | United Kingdom | 18,391 | 4 (<1%) | 1 (<1%) |
| Switzerland | 325 | 4 (1%) | - | |
| Netherlands | 840 | 2 (<1%) | - |
Table 3. Prevalence of SARS-CoV-2 lineages B.1.1.EU/BR and B.1.1.BR across Brazilian states.
|State|Total SARS-Cov-2|Lineage B.1.1.EU/BR|Lineage B.1.1.BR|
|—|—|—|—|—|
|Rio de Janeiro|78|-|59 (76%)|
|Minas Gerais|45|6 (13%)|-|
|Sao Paulo|19 |- |-|
|Distrito Federal|6|1 (17%)| 5 (83%)|
|Amapá|6|-|6 (100%)|
|Pará|6|-|2 (33%)|
|Others|11|-|2 (18%)|
|Total|171|7 (4%)|74 (43%)|
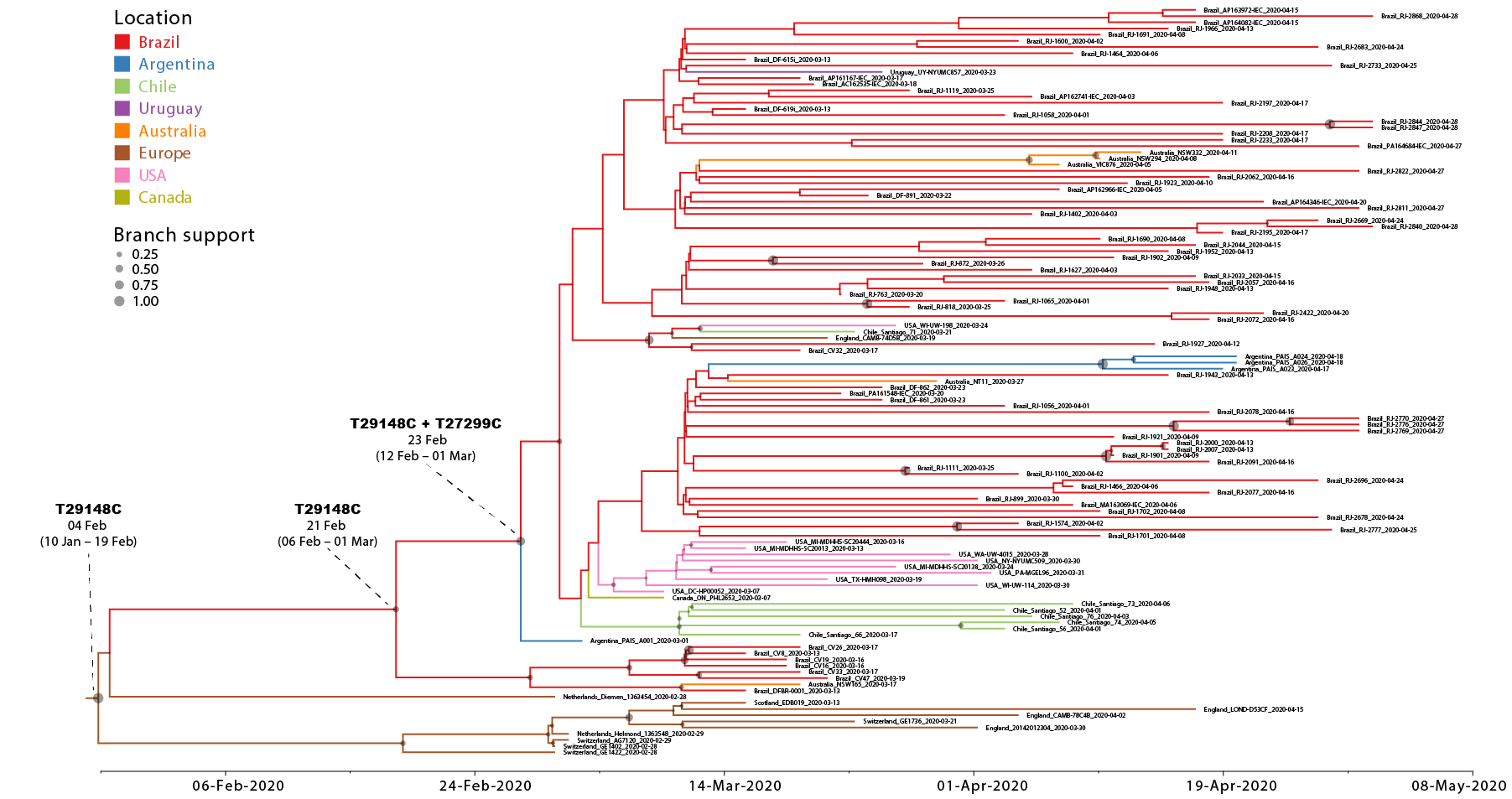
We next conducted a discrete Bayesian phylogeographic analysis to reconstruct the spatiotemporal dissemination dynamics of the B.1.1.EU/BR and B.1.1.BR lineages. Time-scaled trees were estimated by conducted using a strict molecular clock model with fixed substitution rate (8 x 10-4 substitutions/site/year), an HKY+I+G nucleotide substitution model, and the Bayesian skyline coalescent prior as implemented in BEAST 1.10 ( 24 ). Bayesian reconstructions traced the origin of the B.1.1.EU/BR lineage most probably to Europe ( PSP = 0.61) at 4th February (95% High Posterior Density [HPD]: 10th January – 19th February) and the origin of the B.1.1.BR lineage to Brazil ( PSP = 0.95) at 23th February (95% HPD: 12th February – 1st March) (Figure 2). From Brazil, the B.1.1.BR lineage probably disseminated to neighboring South American countries (Argentina, Chile and Uruguay) and to more distant regions (Australia, USA and UK). Although the origin of the B.1.1.BR lineage was placed in Brazil with high probability, the earliest B.1.1.BR sequence currently available corresponds to one Argentinean strain sampled on 1st March 2020. The earliest detection of the B.1.1.BR clade in Brazil occurred in a sample isolated in the Distrito Federal on 13th March 2020. Of note, none of the 30 genomes analyzed between 25th February and 12th March, including seven B.1.1 genomes from imported cases in Sao Paulo, belong to the B.1.1.BR clade.
Although the high-quality full genomes of SARS-CoV-2 currently available contain sufficient phylogenetic information to allow reliable inference of phylogenetic relationships, analyses assessing within-country transmission clusters should be interpreted with caution ( 25 ). The recent emergence and rapid dissemination of the pandemic B.1 clade within and between countries imposes a serious limitation for accurate phylogeographic reconstructions of this SARS-CoV-2 lineage. A test of phylogeny-location association in the posterior sampling of trees obtained from the B.1.1.EU/BR + B.1.1.BR dataset rejected the null hypothesis of panmixia, thus indicating a significant spatial structure in the overall data. Monophyletic clade test, however, revealed random clustering for most of the locations, with the exception of Brazil, Argentina and Europe. Another important limitation is the uneven spatial and temporal sampling. The much higher prevalence of the B.1.1.BR lineage here observed in Brazil relative to North America, Europe or Australia could not be explained by sampling bias, as those regions comprise the most densely sampled countries worldwide. Phylogeographic inference of the origin of the B.1.1-BR lineage, however, will require more sequences from neighboring South American countries, particularly from Argentina, and a denser sampling of the Brazilian epidemic during the very early phase between the late February and early March.
In summary, this study suggests that community transmission of SARS-CoV-2 in Brazil was mainly driven by a single B.1.1 national lineage that probably started to spread in Brazil around 23th February (12th February – 1st March), shortly before the detection of the first imported SARS-CoV-2 case. Continuous efforts for widespread sequencing of SARS-CoV-2 may provide unique insight about the local viral dissemination in Brazil and other South American countries.
References
-
N. Zhu et al. , A Novel Coronavirus from Patients with Pneumonia in China, 2019. N Engl J Med 382 , 727-733 (2020).
-
V. Coronaviridae Study Group of the International Committee on Taxonomy of, The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 5 , 536-544 (2020).
-
E. Dong, H. Du, L. Gardner, An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis 20 , 533-534 (2020).
-
Brazilian Ministry of Health, Brasil confirma primeiro caso da doença – COVID-19. . (2020).
-
J. Hadfield et al. , Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34 , 4121-4123 (2018).
-
Y. Shu, J. McCauley, GISAID: Global initiative on sharing all influenza data - from vision to reality. Euro Surveill 22 , (2017).
-
A. Rambaut et al. , A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. bioRxiv , (2020).
-
R. J. Rockett et al. , Revealing COVID-19 Transmission by SARS-CoV-2 Genome Sequencing and Agent Based Modelling. bioRxiv , (2020).
-
T. Seemann et al. , Tracking the COVID-19 pandemic in Australia using genomics. medRxiv , (2020).
-
S. Dellicour et al. , A phylodynamic workflow to rapidly gain insights into the dispersal history and dynamics of SARS-CoV-2 lineages. bioRxiv , (2020).
-
A. Bluhm et al. , SARS-CoV-2 Transmission Chains from Genetic Data: A Danish Case Study. bioRxiv , (2020).
-
F. Gámbaro et al. , Introductions and early spread of SARS-CoV-2 in France. bioRxiv , (2020).
-
D. F. Gudbjartsson et al. , Spread of SARS-CoV-2 in the Icelandic Population. N Engl J Med , (2020).
-
D. Miller et al. , Full genome viral sequences inform patterns of SARS-CoV-2 spread into and within Israel. medRxiv , (2020).
-
B. B. Oude Munnink et al. , Rapid SARS-CoV-2 whole genome sequencing for informed public health decision making in the Netherlands. bioRxiv , (2020).
-
F. Díez-Fuertes et al. , Phylodynamics of SARS-CoV-2 transmission in Spain. bioRxiv , (2020).
-
A. S. Gonzalez-Reiche et al. , Introductions and early spread of SARS-CoV-2 in the New York City area. Science , (2020).
-
X. Deng et al. , Genomic surveillance reveals multiple introductions of SARS-CoV-2 into Northern California. Science , (2020).
-
M. Worobey et al. , The emergence of SARS-CoV-2 in Europe and the US. bioRxiv , (2020).
-
J. Xavier et al. , The ongoing COVID-19 epidemic in Minas Gerais, Brazil: insights from epidemiological data and SARS-CoV-2 whole genome sequencing. medRxiv , (2020).
-
J. G. Jesus et al. , Importation and early local transmission of COVID-19 in Brazil, 2020. Rev Inst Med Trop Sao Paulo 62 , e30 (2020).
-
P. C. Resende et al. , SARS-CoV-2 genomes recovered by long amplicon tiling multiplex approach using nanopore sequencing and applicable to other sequencing platforms. bioRxiv , (2020).
-
L. T. Nguyen, H. A. Schmidt, A. von Haeseler, B. Q. Minh, IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32 , 268-274 (2015).
-
M. A. Suchard et al. , Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol 4 , vey016 (2018).
-
C. Mavian, S. Marini, M. Prosperi, M. Salemi, A Snapshot of SARS-CoV-2 Genome Availability up to April 2020 and its Implications: Data Analysis. JMIR Public Health Surveill 6 , e19170 (2020).
Acknowledgements
The authors wish to thank all the health care workers and scientists, who have worked hard to deal with this pandemic threat, the Genbank and GISAID team and all the submitters of the database. GISAID acknowledgment tables containing sequences used in this study are attached gisaid_hcov-19_acknowledgement_table_South America.zip (32.2 KB) and gisaid_hcov-19_acknowledgement_table_B.1.1.zip (177.1 KB) ) to this post. Locally, we acknowledge the Respiratory Viruses Genomic Surveillance Network of the General Laboratory Coordination (CGLab) of the Brazilian Ministry of Health (MoH), Brazilian Central Laboratory States (LACENs), and local surveillance teams for the partnership in the viral surveillance in Brazil.
Funding support: CGLab/MoH (General Laboratories Coordination of Brazilian Ministry of Health) and CVSLR/FIOCRUZ (Coordination of Health Surveillance and Reference Laboratories of Oswaldo Cruz Foundation).
Figure 1 . Maximum-likelihood phylogeny of the B.1.1 subclade with the Brazilian strains marked with the red circles and the B.1.1.EU/BR highlighted with the red box (panel A). Panel B shows a magnification of the B.1.1.EU/BR and B.1.1.BR clades. The names of the new Brazilian SARS-CoV-2 genomes generated in this study are in bold.
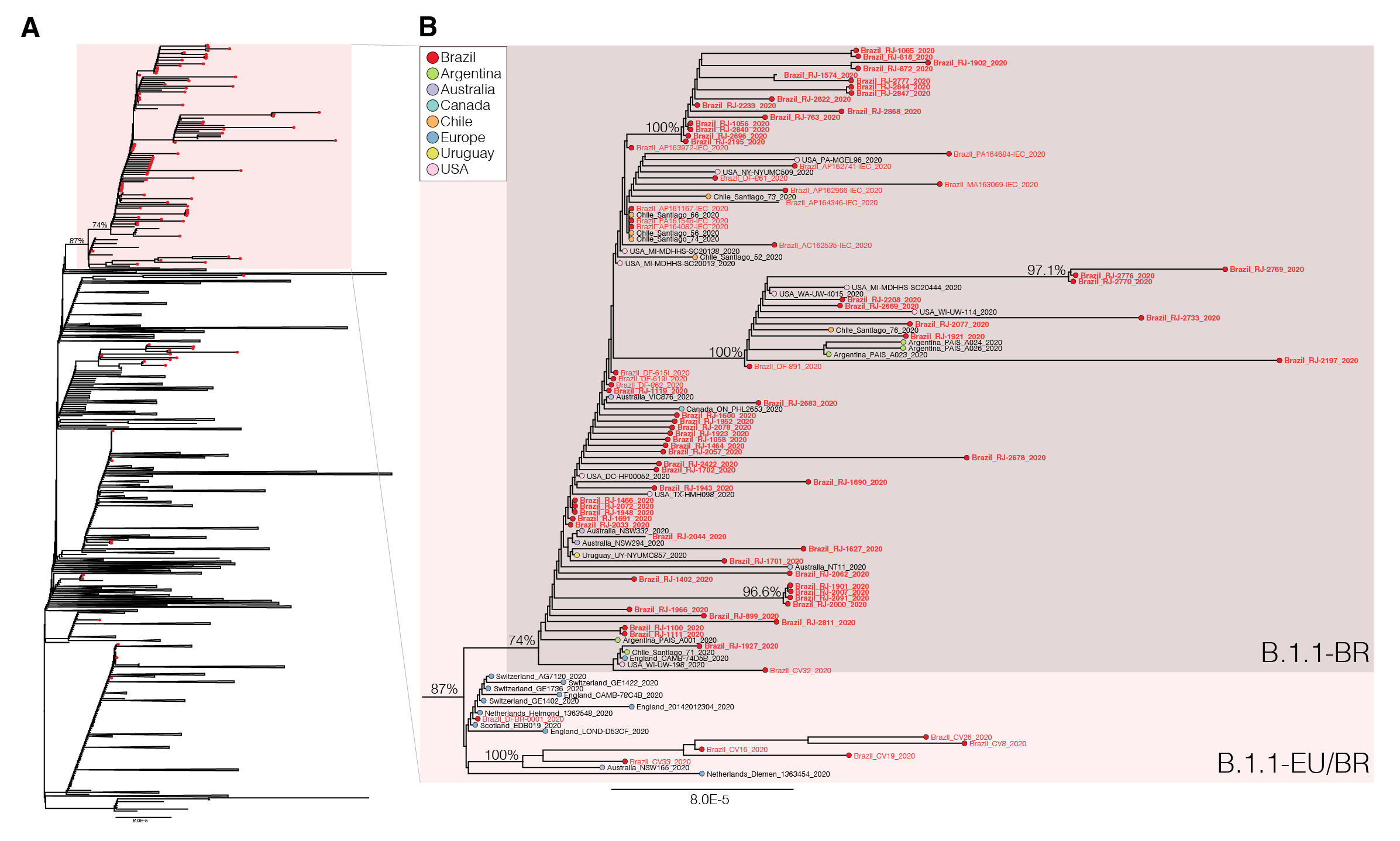
Figure 2. Time-scaled Bayesian phylogeographic MCC tree of the major SARS-CoV-2 lineage circulating in Brazil (B.1.1.BR) and closely related basal strains (B.1.1.EU/BR). Branches are colored according to the most probable location state of their descendent nodes as indicated at the legend. Circles size at internal nodes is proportional to the corresponding posterior probability support as indicated at the legend. The inferred TMRCA (based on the median of the posterior heights) and nucleotide substitutions fixed at ancestral key nodes are shown. The tree is automatically rooted under the assumption of a strict molecular clock and all horizontal branch lengths are drawn to a scale of years.
Supplementary table 1. Epidemiological data of the Brazilian SARS-CoV-2 genomes produced in this study. Suplementary table 1.zip (36.8 KB)