Brazil is experiencing a large outbreak of dengue and severe dengue cases, with 1,127,244 reported between January and June 2019, which corresponds to 88% of all the dengue cases reported in the Americas during this period (1). Within Brazil, São Paulo state notified nearly half of the dengue infections in the country. According to official data from the Ministry of Health, DENV serotype 2 is the dominant strain circulating in 2019 in Brazil (2).
We generated and analysed dengue virus genome data recovered from samples collected between early June 2017 to April 2019 in São José do Rio Preto (SJRP) and Araraquara (ARA), two municipalities in São Paulo state. SJRP samples were obtained from virological routine surveillance, with the study being approved by the University of São José do Rio Preto Institutional Review Board approval #48982/2012. Residual anonymised clinical diagnostic samples from ARA were obtained following ethical approval by Hospital das Clínicas, University of São Paulo’s Institutional Review Board (CAPPesq) (number 3.156.894).
From early June 2017 to April 2019, 734 of 2,793 tested subjects were positive for DENV serotype 2 (DENV2) infection in SJRP; and 400 of 930 tested positive for DENV2 in ARA. Twenty RT-qPCR DENV2 positive samples (mean RT-qPCR cycle threshold: 19.8, range: 16.4 to 25) were randomly selected for complete genome sequencing using a previously developed amplicon-based approach for on-site sequencing using the minION platform (3, 4). Samples were subjected to viral genomic amplification at the Institute of Tropical Medicine, University of São Paulo, Brazil. Genome sequencing was conducted using the handheld nanopore MinION sequencing platform, which has been used previously in Brazil during outbreaks of Zika virus (4, 5), yellow fever virus (6) and chikungunya (7). Sequencing was performed using a multiplex PCR primer scheme designed to amplify the entire coding region of DENV2 as previously described (3).
DENV2 is classified into six genotypes, named I-VI. Up to date, 3 genetic lineages of DENV2 genotype III (DENV2-III) had been reported in Brazil on the basis of phylogenetic analysis of the relationships of partial and complete genomes of circulating strains. Each of these lineages resulted from independent introductions of virus lineages from the Caribbean region in 1990 (DENV2-III lineage 1), 1998 (DENV2-III lineage 2) and 2005 (DENV2-III lineage 3) (8).
To contextualize the diversity of DENV lineages associated with the 2019 explosive epidemic in Brazil, new sequences were appended to a global dataset of 1,630 publicly available DENV2 genomes. Maximum likelihood (ML) phylogenies were generated using PhyML (9) and BEAST 1.10 (10) using a GTR nucleotide substitution model with gamma heterogeneity distributed among site rates. Because we observed that all sequences from the Americas (including the newly generated sequences) grouped together in a well-supported monophyletic clade, we then constructed a dataset comprising only sequences collected in the Americas (n=436).
Our ML phylogenetic analyses indicate that the 2019 sequenced cases belong to DENV2 genotype III (also known as American/Asian genotype). Notably, our analysis indicates that most of the recent DENV2 genomes from São Paulo state (18 of the 19 sequences collected in 2019) cluster together in a single well-supported monophyletic clade (aLTR=1.00), suggesting that this virus lineage results from a novel introduction of a DENV2-III virus lineage from outside of Brazil (named hereafter as DENV2-III lineage 4). The Brazilian DENV2-III lineage 4 is most closely related to publicly available sequences from the Martinique and Guadeloupe.
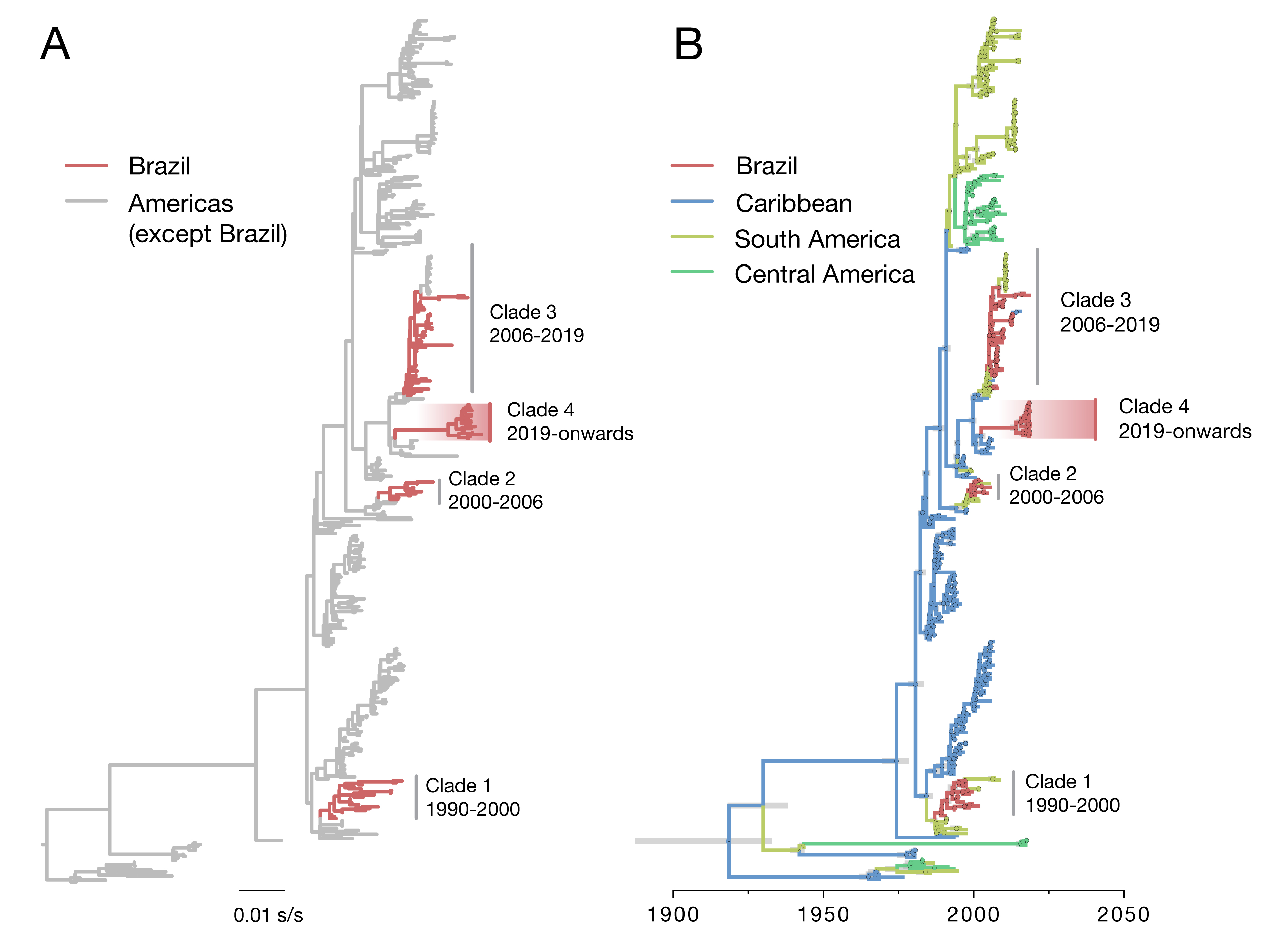
Figure 1. Evolutionary history of DENV2 in Brazil. Maximum likelihood phylogeny (A) and dated phylogeographic tree (B) of DENV serotype 2 ( n =436) in the Americas. Tips (and nodes leading to) Brazilian strains are shown in red. DENV2-III virus lineages (clades) comprising isolates from Brazil are highlighted with a red gradient. In panel B, 95% Bayesian credible intervals for node ages are shown for nodes with Bayesian posterior support above 0.95.
To investigate in more detail the origins of the DENV2-III lineages circulating in Brazil, we conducted a statistical phylogeographic analysis on the 436 DENV2 genome sequences that represent DENV2 diversity in the Americas. Our analysis reveals that the DENV2-III lineage 4 was introduced in or around 2014 (95% Bayesian credible interval: 2012 to 2015). Estimation of its ancestral root location indeed reveals an origin in the Caribbean region (location posterior support = 1.00).
Previous molecular clock analyses have shown that DENV2 lineages have been introduced in Brazil every 7 to 10 years, after which they are replaced by a novel lineage introduced from other locations. Given that the previously circulating DENV2-III lineage 3 had been introduced around 2005 in Brazil, our dating estimates for the introduction of DENV2-III lineage 4 in 2015 is in agreement of a new DENV2 lineage replacement in the country.
Interestingly, two sequences from SJRP, one from June 2017 (ID: 126) and another from January 2019 (ID: 140), grouped together with isolates from the DENV2-III lineage 3 that has been circulating since 2006 in the Southeast Region. This suggests co-circulation of two DENV2-III lineages (lineages 3 and 4) in a single location (SJRP). According to our yet limited genomic dataset from the ongoing outbreak, the frequency of lineage 3 in SJRP is 5.3% (n=1/19) and 94.7% for lineage 4 (n=18/19). However, the timing of the introduction of the DENV2-III lineage 4, the explosive epidemic associated with the high increase in the number of dengue cases reported in São Paulo state, combined with the increasing frequency of lineage 4 in relation to lineage 3 suggests that we are capturing the replacement of DENV2 virus lineages in real-time.
The temporal lag between the detection and the estimated date of introduction of the DENV2-III lineage 4 highlight the need of improved surveillance in areas where virus lineages might circulate undetected. For DENV, descriptive reporting (e.g. 11) and genomic analyses (12, 13) suggest that the north Amazon region of Brazil may act an epidemiological bridge for DENV movement from regions with year-round DENV transmission in Aedes spp. mosquitoes (such as the Caribbean region) into the rest of Brazil. Integration of routine serological, molecular, and genomic data along with improved digital surveillance (14) will help to anticipate the arrival and establishment of new virus lineages and other pathogens across different regions in Brazil.
Authors
Jaqueline Goes de Jesus
Karina Rocha Dutra
Flavia Cristina da Silva Salles
Ingra Morales Claro
Ana Carolina Terzian
Darlan da Silva Candido
Sarah C. Hill
Julien Thézé
Alvina Clara Felix
Andreia F. Negri Reis
Luiz Carlos Junior Alcantara
André L. Abreu
Júlio H. R. Croda
Wanderson K. de Oliveira
Ana Maria Bispo de Filipis
Camila Malta Romano
Nick J. Loman
Oliver G. Pybus
Ester Cerdeira Sabino
Mauricio L. Nogueira
Nuno Rodrigues Faria
Affiliation
Instituto de Medicina Tropical, Universidade de São Paulo, São Paulo, Brazil.
Laboratório de Pesquisa em Virologia, Faculdade de Medicina de São José do Rio Preto, São José do Rio Preto, Brasil.
Department of Zoology, University of Oxford, South Parks Road, Oxford, UK.
Secretaria Municipal de Saúde, São José do Rio Preto, São Paulo, Brazil.
Laboratório de Flavivírus, Instituto Oswaldo Cruz, FIOCRUZ, Rio de Janeiro, Brazil.
Universidade Federal de Minas Gerais, Belo Horizonte, Brazil.
Secretaria de Vigilância em Saúde, Coordenação Geral de Laboratórios de Saúde Pública, Ministério da Saúde, Brasília-DF, Brazil.
Laboratório de Pesquisa em Ciências da Saúde, Universidade Federal da Grande Dourados, Dourados, Mato Grosso do Sul, Brazil.
Fundação Osvaldo Cruz Campo Grande, Mato Grosso do Sul, Brazil.
Hospital das Clínicas HCFMUSP (LIM52), Faculdade de Medicina, Universidade de São Paulo, São Paulo, Brazil.
Hospital das Clínicas HCFMUSP (LIM52), Faculdade de Medicina, Universidade de São Paulo, São Paulo, Brazil.
Funding: The research was supported by the FAPESP-MRC grant (CADDE, FAPESP 2018/14389-0), a Wellcome Trust and Royal Society Sir Henry Dale Fellowship (grant 204311/Z/16/Z), by a CNPq # 400354/2016-0 and FAPESP # 2016/01735-2, and by the Oxford Martin School. This work was supported by Decit/SCTIE/MoH and CNPq (440685/2016-8 and 440856/2016-7); by CAPES (88887.130716/2016-00, 88881.130825/2016-00 and 88887.130823/2016-00); by EU’s Horizon 2020 Programme through ZIKAlliance (PRES-005-FEX-17-4-2-33). MLN is supported by FAPESP (Grant # 13/21719-3). ACBT and KRD receive FAPESP Scholarships (Grants #15/12295-0 and 15/14313-6, respectively). MLN and ECS are CNPq Research Fellows (PQ).
Disclosure and contact details: The results presented here by the CADDE project (Brazil-UK Centre for Arbovirus Discovery, Diagnostics, Genomics and Epidemiology) team should be considered preliminary. For additional information please contact us.