Sarah C Hill 1⤉*, Nikita S. D. Sahadeo 2⤉, Lana Gyan 3, Vernie Ramkissoon 2, Rod Suepaul 4, Gabriel Gonzalez-Escobar 5, Saed Rahaman 6, Christopher Oura 4, Christine V. F. Carrington 2*.
1 The Royal Veterinary College, UK; 2 Department of Preclinical Sciences, Faculty of Medical Sciences, The University of the West Indies, St. Augustine, Trinidad and Tobago; 3 Veterinary Diagnostic Laboratory, Ministry of Agriculture, Land and Fisheries, Trinidad and Tobago; 4 School of Veterinary Medicine, Faculty of Medical Sciences, The University of the West Indies, St. Augustine, Trinidad and Tobago; 5 Caribbean Public Health Agency, Federation Park, Trinidad and Tobago; 6 Veterinary Public Health Unit, Ministry of Health, Trinidad and Tobago.
⤉ These authors contributed equally to this work.
* Corresponding authors
Summary: A non-human primate (red howler monkey; Alouatta seniculus) was found positive for yellow fever virus (YFV) in Trinidad in February 2021. This represented the first detection of YFV in the country since 2009. Rapid sequencing of the virus genome at The University of the West Indies demonstrated that this outbreak was caused by a different lineage to the 2009 outbreak.
On February 8th 2021 an adult, female red howler monkey (Alouatta seniculus) carcass was identified and collected by The Veterinary Public Health Unit (VPHU) of the Ministry of Health, Republic of Trinidad and Tobago, during their routine YFV surveillance of forested areas. The carcass was one of two Red Howler monkeys found dead in the Brigand Hill area of Plum Mitan, near the east-coast of the island of Trinidad (co-ordinates: 10.488351, 61.085938). During their visit to the area, the VPHU noted the presence of mosquitoes, as well as two red howler monkey troops with no obvious signs of illness in those live monkeys visually observed. The carcass, which was intact and had no signs of open wounds, was brought to the Veterinary Diagnostic Laboratory of the Ministry of Agriculture, Land and Fisheries, where a post-mortem was conducted on February 10th 2021.
Upon post-mortem the carcass was found to be in a moderate state of autolysis and in good body condition with adequate subcutaneous and visceral fat stores.The carcass was mildly dehydrated. The liver was diffusely yellow with multifocal, circular (1-2 mm in diameter) dark-red lesions scattered on the capsular surface of all liver lobes, as well as the capsular surface of the kidney. Splenomegaly was also observed. All other body systems examined during the post-mortem were found to be within normal gross limits. The histopathological diagnosis was moderate hepatitis, mixed cellular, with mid-zonal necrosis and diffuse hepatic lipidosis. These findings were thought to be suspicious of YFV infection and tissue samples (liver, kidney and spleen) were sent to the Caribbean Public Health Agency for confirmatory molecular testing. Results of real-time PCR indicated that all three tissues were positive for YFV, with PCR Cts in the range of 8.54 – 15.80.
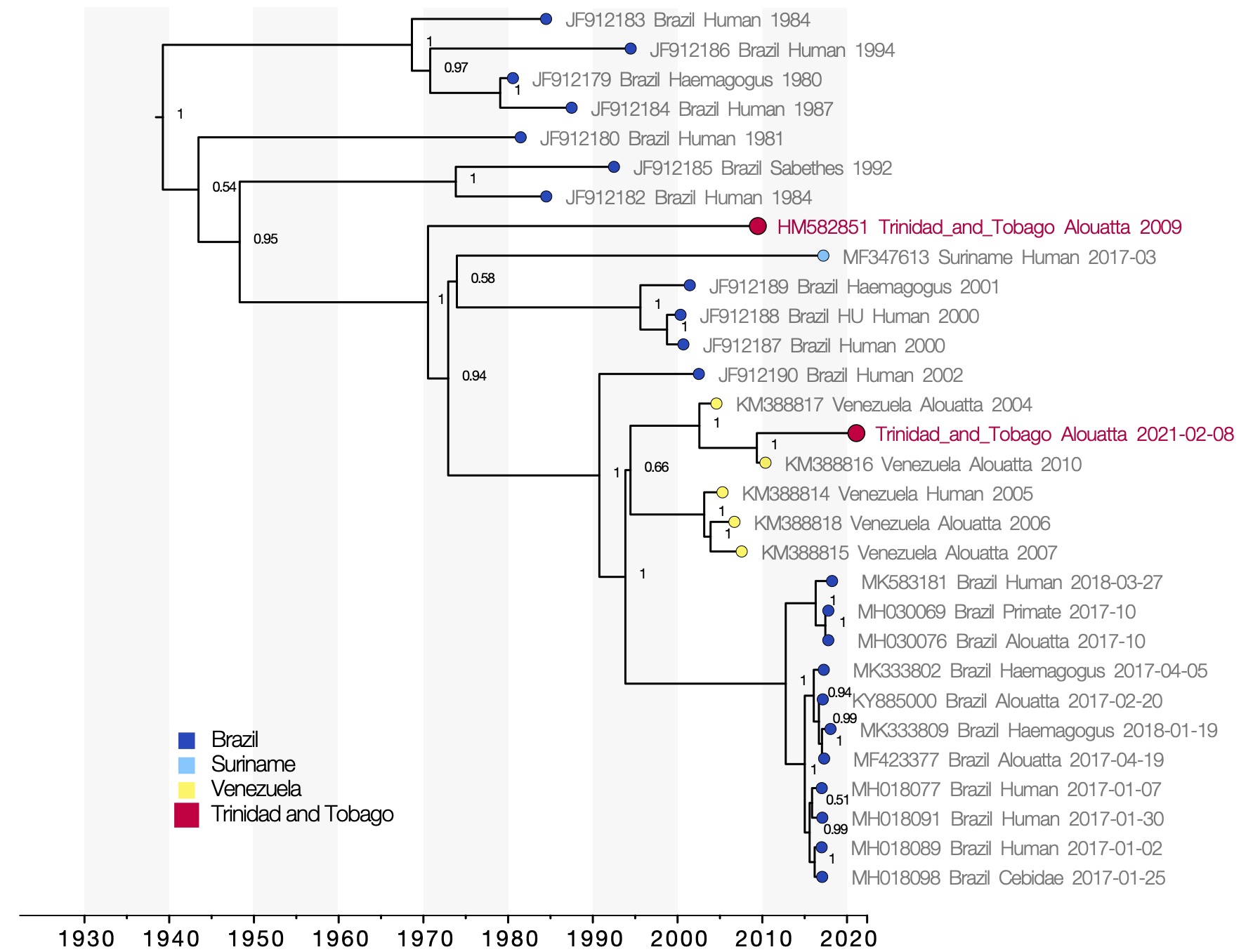
Figure 1 : Molecular clock phylogeny. Viruses sequenced from the 2021 and 2009 outbreaks of yellow fever virus in Trinidad and Tobago are marked in red. Numbers at the nodes indicate posterior support for clade structure. Tips colours indicate the country of sampling of each virus. The x-axis shows the time at which reconstructed common ancestors are estimated to have occurred.
The UWI generated a near complete genome sequence (97% of the protein coding region) using pooled amplicons from the YFV positive kidney and liver samples (including all reads from these tissues). The genome belongs to the South American I genotype of YFV. This is the second complete YFV genome to be reported from Trinidad and Tobago.
YFV genomes from the 2021 and the 2009 outbreaks in Trinidad fall in different positions in the phylogenetic tree (Figure 1), indicating that the outbreaks are not directly related and each represent separate introductions of YFV to Trinidad. The 2021 Trinidad YFV isolate shares a most recent common ancestor with an isolate sampled in Venezuela in 2010 with strong posterior support (1). This relationship does not change in analyses considering only partial gene regions, which allow a greater number of short sequences from public databases to be considered (not shown here). The most recent common ancestor of the 2010 YFV isolate from Venezuela and the 2021 isolate from Trinidad is dated to May 2009 (95% highest posterior density interval, August 2007 to August 2010). The YFV lineage may therefore have been introduced to Trinidad any time between 2007 and 2021.
Given that continuous YFV surveillance in Trinidad has not yielded positive detections since 2009, we speculate that the lineage may have been introduced relatively recently within that time period. The virus may have been introduced from Venezuela, although the long branch (12 years) and lack of virus genomic sequences from other outbreaks that have occurred in mainland South America more recently than the 2010 Venezuelan outbreak (e.g., including Colombia, French Guiana, Venezuela amongst others) make it difficult to be fully confident about the country from which the virus was imported.
The current re-emergence of YFV in Trinidad and Tobago is not unexpected. YFV has been introduced to Trinidad at least twice previously over the past forty years (Auguste et al, 2010), and epizootics in Trinidad and Tobago occur on average every 10 years (1979, 1988-1989, 1995, 2008-2009, and now 2021) (Auguste et al, 2010). That YFV has been able to repeatedly disperse across the ocean to Trinidad has implications for our broader understanding of YFV spatial dispersal. Specifically, the involvement of ‘bridge vectors’ is not necessarily required to explain land-based dispersal of sylvatic outbreaks across areas that lack suitability for yellow fever virus mosquitos or non-human primates. YFV movement between unconnected patches with high-suitability may therefore occur regularly in sylvatic outbreaks. Wind dispersal of mosquitos, human migration and illegal importation of animals into Trinidad from the mainland all remain possible entry routes for the virus.
Methods:
Ribonucleic acid (RNA) samples extracted were extracted from kidney, liver and spleen samples collected from the carcass using Qiagen Viral RNA mini kits. These samples were tested for YFV by RT-PCR (Domingo et al, 2012) by the Caribbean Public Health Agency. RNA samples were subjected to reverse transcription and PCR amplified using a set of previously published YFV specific primers spanning the full genome (Faria et al, 2018). The pooled amplicons for each sample were subjected to gel electrophoresis using a 2% agarose gel. Pooled amplicons from the kidney and liver samples were converted to libraries using the LoCost protocol described for SARS-CoV-2 sequencing by the ARTIC Network (Quick et al, 2020, Tyson et al, 2020). Libraries were sequenced on an Oxford Nanopore Technologies MinION, using a R10.3 flowcell and the MinKNOW software (v20.03.6; Oxford Nanopore Technologies). The fast5 files containing raw reads were basecalled using Guppy (v3.6.0; Oxford Nanopore Technologies). Variant calling and final sequence assembly was carried out using Medaka, with a 2002 Brazilian sequence (Genbank Accession Number JF912190) used as the reference.
The generated YFV genome from Trinidad was aligned with other whole genome YFV South American I genotype sequences (>70% length) from GenBank. Sequences from the Brazilian 2017-2019 YFV outbreak were randomly downsampled to 10 genomes because these sequences were heavily overrepresented in the dataset, yielding a final dataset of 30 genomes. A molecular clock phylogeny was estimated using BEAST v1.10.4, using a Skygrid coalescent model with 30 groups, a relaxed clock with lognormally-distributed rate heterogeneity, and a general-time reversible substitution model with gamma-distributed among-site rate variation and invariant sites. Tree files produced through two independent MCMC chains were combined, each with 20 million steps.
Data availability:
This post presents preliminary findings at an early stage in the investigation into the 2021 Trinidad outbreak, and will be updated if additional genomic or epidemiological findings become available to us. We are very happy to share the virus genomic sequence for use in other studies before it is publicly available on GenBank; please contact Christine Carrington at The UWI to arrange [email protected].
Acknowledgements:
NS is funded by a Trinidad and Tobago-University of the West Indies Research Development Impact Fund Innovation Proof of Concept Grant (grant 26607-447524) to CVFC. SCH is funded by the Wellcome Trust (grant 220414/Z/20/Z). We thank Flávia Salles (IMT, University of São Paulo) and the CADDE Project for kindly sharing PCR primers for genome sequencing.
References
Auguste AJ et al. Yellow fever virus maintenance in Trinidad and its dispersal throughout the Americas. J Virol. 2010 Oct;84(19):9967-77. doi: 10.1128/JVI.00588-10. Epub 2010 Jul 14. PMID: 20631128; PMCID: PMC2937779.
Domingo C et al. J Clin Microbiol. 2012 Dec;50(12):4054-60. doi: 10.1128/JCM.01799-12. Epub 2012 Oct 10. PMID: 23052311; PMCID: PMC3503008.
Faria NR et al. Genomic and epidemiological monitoring of yellow fever virus transmission potential. Science. 2018 Aug 31;361(6405):894-899. doi: 10.1126/science.aat7115. Epub 2018 Aug 23. PMID: 30139911; PMCID: PMC6874500.
Quick J. nCoV-2019 sequencing protocol v3 (LoCost) (protocols.io) Protocols.io 2020
Tyson et al. Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. bioRxiv [Preprint]. 2020 Sep 4:2020.09.04.283077. doi: 10.1101/2020.09.04.283077. PMID: 32908977; PMCID: PMC7480024.